第 3 章
解释 1
这段内容是文档的最高级标题,标志着第三章的开始。本章主要围绕有机化学的基础——烃类化合物展开。内容将包括官能团的介绍、烃的系统分类方法(如饱和/不饱和、环状/无环等)、同分异构体的概念(特别是构造异构体)、国际纯化学和应用化学联合会(IUPAC)的系统命名法,以及烷烃分子内部因单键旋转而产生的不同空间排布,即构象分析。
表 3.1 列出了官能团。可作参考 - 无需记忆。
解释 2
该段落是一条提示性说明,告知读者表3.1包含了“官能团”的列表。官能团(Functional Group)是有机分子中决定其化学性质特征的原子或原子团。例如,羟基(-OH)是醇的官能团,羧基(-COOH)是羧酸的官能团。这里特别指出,这个表格仅供参考,现阶段不需要强行记忆所有官能团,暗示了在后续学习中会逐步接触和熟悉它们。
1 烃的分类hydrocarbons=CH
解释 3
这是一个一级标题,标题为“烃的分类”。“Hydrocarbons=CH”是一个简明的注解,说明烃(Hydrocarbons)是由碳(C)和氢(H)两种元素组成的有机化合物。本节将详细介绍对这类化合物进行分类的不同标准和术语。
饱和烃saturated:仅含单键(无 键)
解释 4
该段落定义了“饱和烃”(Saturated Hydrocarbons)。 核心定义:饱和烃是指分子中所有的碳-碳键均为单键的烃类化合物。 详细解释:
- 单键:碳原子之间通过共用一对电子形成的化学键,称为(西格玛)键。这种键连接牢固,可以自由旋转。
- “饱和”的含义:在饱和烃中,每个碳原子都已经与最大可能数目的原子(氢原子或其他碳原子)成键。因此,它们不能再通过加成反应与其它原子(如氢)结合,其碳骨架已经达到了“氢饱和”的状态。
- 无键:双键由一个键和一个(派)键组成,三键由一个键和两个键组成。由于饱和烃只含单键,所以它们分子中不存在键。 举例说明:
- 乙烷(Ethane),化学式为(结构简式为),分子中只含有碳-碳单键和碳-氢单键,因此是典型的饱和烃。
- 环己烷(Cyclohexane),化学式为,虽然是环状结构,但其环上的碳原子之间也全部是单键,所以也属于饱和烃。
不饱和烃unsaturated:含 键
解释 5
该段落定义了“不饱和烃”(Unsaturated Hydrocarbons)。 核心定义:不饱和烃是指分子中含有至少一个碳-碳双键或碳-碳三键的烃类化合物。 详细解释:
- 含键:如前所述,双键和三键中都包含键。键的电子云分布在键的上方和下方,相对活泼,容易断裂,使不饱和烃能够发生加成反应。
- “不饱和”的含义:与饱和烃相比,不饱和烃分子中的氢原子数目没有达到最大值。它们可以通过打开键,与氢气等试剂发生加成反应,转变为饱和烃。例如,乙烯可以与氢气反应生成乙烷。 举例说明:
- 乙烯(Ethene),化学式为(结构简式为),含有一个碳-碳双键,属于不饱和烃。
- 乙炔(Ethyne),化学式为(结构简式为),含有一个碳-碳三键,也属于不饱和烃。
环状烃cyclic:含一个或多个环
解释 6
该段落定义了“环状烃”(Cyclic Hydrocarbons)。 核心定义:环状烃是指分子中的碳原子通过化学键形成一个或多个闭合环状结构的烃类化合物。 详细解释:
- 环状烃的碳原子骨架不是开放的链状,而是首尾相连形成环。
- 环状烃既可以是饱和的(如环烷烃,环中只有单键),也可以是不饱和的(如环烯烃,环中有双键)。 举例说明:
- 环丙烷(Cyclopropane),化学式为,是分子中含有三元碳环的饱和环状烃。
- 苯(Benzene),化学式为,是含有六元碳环的特殊不饱和环状烃(芳香烃)。
无环烃acyclic:不含环
解释 7
该段落定义了“无环烃”(Acyclic Hydrocarbons)。 核心定义:无环烃是指分子中的碳原子连接成链状结构,而不形成任何环的烃类化合物。这类烃也被称为链烃。 详细解释:
- 无环烃的碳链可以是直链(所有碳原子排成一条线)或支链(主链上连接有侧链)。
- 与环状烃相对,它们是开放式的结构。 举例说明:
- 正丁烷(Butane),化学式为(结构简式为),是一个直链无环烃。
- 异丁烷(Isobutane),化学式也是(结构简式为),是一个支链无环烃。
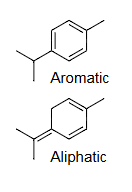
芳香烃aromatic:含苯环benzene或类似结构analogous
解释 8
该段落定义了“芳香烃”(Aromatic Hydrocarbons)。 核心定义:芳香烃是一类特殊的环状不饱和烃,其分子中含有苯环(Benzene ring)或结构与性质类似于苯环的体系。 详细解释:
- 苯环:苯()是一个六元碳环,环内的6个电子形成一个离域大键,使得苯环具有特殊的稳定性,这种性质称为“芳香性”。
- 类似结构:除了苯环,还有其他符合特定规则(如休克尔规则)的环状共轭体系也具有芳香性,例如萘(Naphthalene)。
- 化学性质:芳香烃的化学性质与普通的烯烃、炔烃有很大区别,它们不容易发生加成反应,而倾向于发生取代反应。 举例说明:
- 苯(Benzene, )是最简单、最典型的芳香烃。
- 甲苯(Toluene, ),是苯环上的一个氢原子被甲基()取代后形成的化合物。
脂肪族aliphatic:非芳香族aromatic=可以饱和saturated的或不饱和unsaturated
解释 9
该段落定义了“脂肪族烃”(Aliphatic Hydrocarbons)。 核心定义:脂肪族烃是有机化学中一个非常广泛的分类,它包含了所有非芳香族的烃类化合物。 详细解释:
- 这个术语源于早期化学家发现这类化合物与脂肪(fats)有关。
- 它是一个与“芳香族”相对立的概念。只要一个烃不具备芳香性,它就属于脂肪族烃。
- 因此,脂肪族烃可以是:
- 饱和的:如烷烃(alkanes)。
- 不饱和的:如烯烃(alkenes)和炔烃(alkynes)。
- 无环的(链状的):如正己烷。
- 环状的(非芳香环):如环己烷。 举例说明:
- 所有前面提到的烷烃、烯烃、炔烃、环烷烃、环烯烃都属于脂肪族烃。例如,乙烷(饱和无环)、乙烯(不饱和无环)、环己烷(饱和环状)都是脂肪族烃。
解释 10
这是一张图片,它以流程图的形式直观地总结了烃的分类体系。 图片内容解析: 该图展示了烃(Hydrocarbons)如何被逐级细分:
- 第一级分类:将烃分为两大类:脂肪族烃(Aliphatic) 和 芳香族烃(Aromatic)。
- 第二级分类(脂肪族内部):将脂肪族烃进一步分为 无环烃(Acyclic) 和 脂环烃(Alicyclic,即非芳香的环状烃)。
- 第三级分类(按饱和度):
- 在 无环烃 类别下,根据是否含有键,分为 烷烃(Alkanes,饱和) 和 不饱和烃。不饱和烃又分为 烯烃(Alkenes,含双键) 和 炔烃(Alkynes,含三键)。
- 在 脂环烃 类别下,同样根据饱和度分为 环烷烃(Cycloalkanes,饱和) 和 环烯烃(Cycloalkenes,不饱和)。
这张图提供了一个清晰的知识框架,帮助理解各种烃类术语之间的层级和并列关系。
IUPAC 分类
解释 11
这是一个二级标题,标题为“IUPAC 分类”。IUPAC 是“国际纯化学和应用化学联合会”(International Union of Pure and Applied Chemistry)的缩写。该组织制定了一套全球通用的、系统的化学物质命名规则,即IUPAC命名法。本节将根据IUPAC的体系,对几类基本的烃进行定义。
烷烃alkanes:饱和的。如果是无环的,其分子式为
解释 12
该段落定义了“烷烃”(Alkanes)。 核心定义:烷烃是饱和烃,即分子中只含有碳-碳单键和碳-氢单键的烃。 涉及的公式:
- 无环烷烃的通式:$$ \mathrm{C}{n} \mathrm{H}{2 n+2} $$ 公式详解:
- 代表碳元素, 代表氢元素。
- 是一个正整数,代表分子中碳原子的数目。
- 表示当碳原子数为 时,对应的氢原子数目。这个公式表明,在任何一个无环烷烃分子中,氢原子的数量总是碳原子数量的两倍再加二。 举例说明:
- 当 时,分子式为 ,这是甲烷(Methane)。
- 当 时,分子式为 ,这是丙烷(Propane)。
- 当 时,分子式为 ,这是戊烷(Pentane)。 这个通式是判断一个无环烃是否为烷烃的快速方法。
烯烃alkenes:含 个双键
解释 13
该段落定义了“烯烃”(Alkenes)。 核心定义:烯烃是含有至少一个碳-碳双键()的不饱和烃。 详细解释:
- “”表示“大于或等于1”,即分子中可以有一个或多个双键。
- 由于双键的存在,烯烃属于不饱和烃。
- 含有一个双键的无环烯烃的通式为 (其中 )。 举例说明:
- 乙烯(Ethene),化学式为 ,结构简式为 ,是最简单的烯烃。
- 丙烯(Propene),化学式为 ,结构简式为 。
炔烃alkynes:含 个三键
解释 14
该段落定义了“炔烃”(Alkynes)。 核心定义:炔烃是含有至少一个碳-碳三键()的不饱和烃。 详细解释:
- “”表示“大于或等于1”,即分子中可以有一个或多个三键。
- 由于三键的存在,炔烃也属于不饱和烃,且不饱和度高于对应的烯烃。
- 含有一个三键的无环炔烃的通式为 (其中 )。 举例说明:
- 乙炔(Ethyne),化学式为 ,结构简式为 ,是最简单的炔烃。
- 丙炔(Propyne),化学式为 ,结构简式为 。
2同分异构体Isomers
解释 15
这是一个一级标题,标题为“同分异构体”(Isomers)。本节将介绍有机化学中一个非常重要的概念——同分异构现象。
同分异构体Isomers:分子式相同但结构不同的分子。
解释 16
该段落给出了“同分异构体”(Isomers)的精确定义。 核心定义:同分异构体是指具有完全相同的分子式(即各种元素的原子数量相同),但原子在空间中的排列方式或连接顺序不同的化合物。 详细解释:
- 分子式相同:这是成为同分异构体的前提条件。例如,两个化合物如果分子式都是 ,它们就有可能是同分异构体。
- 结构不同:这是同分异构体的本质特征。结构的不同可以体现在原子的连接顺序上(构造异构),也可以体现在原子在空间中的三维排布上(立体异构)。
- 性质不同:由于结构决定性质,同分异构体虽然分子式相同,但它们的物理性质(如熔点、沸点、密度)和化学性质通常是不同的。 举例说明:
- 乙醇()和甲醚()的分子式都是 ,但它们的结构和性质迥异,是一对同分异构体。乙醇是液体,能与水混溶;甲醚是气体,微溶于水。
构造异构体Constitutional isomers在键连接性bond connectivity上有所不同。
解释 17
该段落定义了同分异构体中的一类——“构造异构体”(Constitutional isomers),也称为“结构异构体”(Structural isomers)。 核心定义:构造异构体是指分子式相同,但分子中原子的连接顺序和方式不同的异构体。 详细解释:
- 键连接性(Bond Connectivity):指的是哪个原子与哪个原子通过化学键相连。构造异构体的根本区别就在于这种连接的“拓扑结构”不同。
- 构造异构可以进一步分为三类:
- 碳链异构:因碳原子的骨架排列不同而产生,如直链与支链。
- 位置异构:因官能团或取代基在碳链上的位置不同而产生。
- 官能团异构:因分子式相同但官能团类型不同而产生。 举例说明:
- 丁烷()和异丁烷()都具有分子式 。在丁烷中,碳原子是线性连接的;而在异丁烷中,存在一个支链。它们的原子连接顺序不同,因此是一对构造异构体(属于碳链异构)。
构造异构体Constitutional isomers
解释 18
这是一个二级标题,指出本节将以分子式为 的化合物为例,具体展示构造异构体。 涉及的公式:
- 分子式:$$ \mathrm{C}{5} \mathrm{H}{12} $$ 公式详解:
- 该分子式表示这个分子由5个碳原子和12个氢原子构成。
- 根据烷烃通式 进行验证,当 时,氢原子数为 。因此, 是一种烷烃,称为戊烷(Pentane)。接下来将展示戊烷存在哪些不同的碳链连接方式。
解释 19
这张图片展示了分子式为 的三种构造异构体的球棍模型和结构式。 图片内容解析: 从左到右,图中列出了三种不同的分子结构,它们都符合 的分子式:
- 正戊烷 (Pentane / n-Pentane):这是一个直链烷烃,5个碳原子线性排列。结构简式为 。
- 异戊烷 (Isopentane):这是一个支链烷烃,主链有4个碳原子,在第二个碳原子上连接了一个甲基支链。其系统命名为“2-甲基丁烷”。结构简式为 。
- 新戊烷 (Neopentane):这是一个高度支链化的烷烃,主链有3个碳原子,在中间的碳原子上连接了两个甲基支链。其系统命名为“2,2-二甲基丙烷”。结构简式为 。
这三者是典型的碳链异构体,因为它们的区别在于碳骨架的结构不同(分别是直链、单支链、双支链)。
直链
解释 20
“直链”这个词和随后的图片是对前述异构体中第一种情况的强调。 图片内容解析: 这张图片特写了正戊烷 (n-Pentane) 的结构。
- 直链 (Straight-chain):这个术语描述的是一种碳骨架结构,其中碳原子一个接一个地连接,形成一条没有分支的链。需要注意的是,“直链”并不意味着分子在三维空间中是笔直的,由于碳-碳单键的键角(约109.5°),碳链实际上是锯齿状的。但从连接性上看,它没有分岔。
支链
解释 21
“支链”这个词指的是除直链结构外的其他碳链结构。 核心定义:
- 支链 (Branched-chain):指碳链中除了首尾两个端点碳原子外,至少有一个碳原子连接了两个以上的其他碳原子,从而在主碳链上形成了“分岔”或“侧链”。 举例说明:
- 前面提到的异戊烷和新戊烷都是支链烷烃的例子,它们分别含有一个和两个支链。
IUPAC 命名法。nomenclature
解释 22
这是一个二级标题,引入了“IUPAC命名法”(IUPAC nomenclature)的讨论。本节将系统介绍如何根据IUPAC制定的一套严谨规则,为有机化合物(此处主要指烷烃及其衍生物)赋予一个唯一的、无歧义的名称。
(在表 3.3 中,记住 的名称)
解释 23
这是一条学习指令,要求学生记住表3.3中从含有1个碳原子()到含有13个碳原子()的直链烷烃的名称。这些名称是构建所有烷烃及其衍生物系统名称的基础词根,因此熟练掌握它们至关重要。例如,记住“meth-”代表1个碳,“eth-”代表2个碳,“prop-”代表3个碳等。
| C(n) | Name | Formula (CₙH₂ₙ₊₂) |
|---|---|---|
| 1 | Methane | CH₄ |
| 2 | Ethane | C₂H₆ |
| 3 | Propane | C₃H₈ |
| 4 | Butane | C₄H₁₀ |
| 5 | Pentane | C₅H₁₂ |
| 6 | Hexane | C₆H₁₄ |
| 7 | Heptane | C₇H₁₆ |
| 8 | Octane | C₈H₁₈ |
| 9 | Nonane | C₉H₂₀ |
| 10 | Decane | C₁₀H₂₂ |
| 11 | Undecane | C₁₁H₂₄ |
| 12 | Dodecane | C₁₂H₂₆ |
| 13 | Tridecane | C₁₃H₂₈ |
| 20 | Icosane | C₂₀H₄₂ |
| 30 | Triacontane | C₃₀H₆₂ |
解释 24
这个表格列出了一系列直链烷烃的名称和分子式,它们是IUPAC命名法的基础。 表格内容解析:
- 第一列 C(n):代表烷烃分子中的碳原子数 。
- 第二列 Name:对应碳原子数的烷烃的英文名称。前四个(Methane, Ethane, Propane, Butane)是习惯用名,从第五个(Pentane)开始,名称的词根源自表示数字的希腊文或拉丁文(penta-=5, hexa-=6, hepta-=7等),后缀统一为“-ane”,表示这是烷烃。
- 第三列 Formula ():给出了对应烷烃的分子式。 涉及的公式:
- 烷烃通式:$$ C_nH_{2n+2} $$ 公式详解:表格中的所有分子式都严格遵循这个通式。例如,对于壬烷(Nonane),,其分子式为 。对于二十烷(Icosane),,其分子式为 。 学习要点:根据前面的提示,需要熟记 到 的烷烃名称,因为它们是确定有机物主链名称的基础。
| 烷烃 | 烷基 | |||
|---|---|---|---|---|
| Alkane | Alk ane | Alkyl | Alk yl | Abbreviation |
| CH₄ | methane | CH₃ | methyl | Me |
| C₂H₆ | ethane | C₂H₅ | ethyl | Et |
| C₃H₈ | propane | C₃H₇ | propyl | Pr |
| C₄H₁₀ | butane | C₄H₉ | butyl | Bu |
解释 25
这个表格展示了如何从烷烃(Alkane)得到对应的烷基(Alkyl group)。 核心概念:烷基是一个从烷烃分子中去掉一个氢原子后形成的官能团或分子片段。烷基不能独立稳定存在,它总是作为分子的一部分,连接在其他原子或基团上。 表格内容解析:
- Alkane -> Alkyl:命名规则是将烷烃名称的后缀“-ane”改为“-yl”。例如,methane 变成 methyl,ethane 变成 ethyl。
- CH₄ (methane) -> CH₃ (methyl):甲烷分子去掉一个氢原子,形成甲基。
- C₂H₆ (ethane) -> C₂H₅ (ethyl):乙烷分子去掉一个氢原子,形成乙基。
- Abbreviation:列出了常用烷基的缩写。在书写复杂的化学结构式时,使用这些缩写可以大大简化表达。
- Me: 甲基 (Methyl)
- Et: 乙基 (Ethyl)
- Pr: 丙基 (Propyl)
- Bu: 丁基 (Butyl) 举例说明:
- 甲苯(Toluene)的结构是一个苯环连接一个甲基,可以写作 Ph-Me(其中Ph代表苯基)。
- 乙酸乙酯(Ethyl acetate)的结构中含有一个乙基,其化学式可以简写为 。
简单的烷基C3H7:
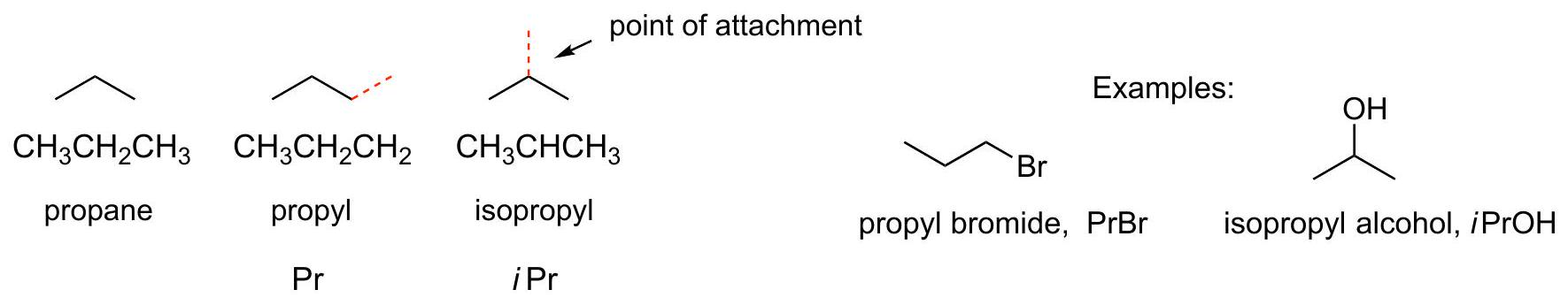
CH₃CH₂CH₃= Propane
CH₃CH₂CH₂–= Propyl=Pr
(CH₃)₂CH–=Isopropyl=iPr
CH₃CH₂CH₂–Br=Propyl bromide=PrBr
(CH₃)₂CH–OH=Isopropyl alcohol=iPrOH
C3=prop=CH3-CH2-CH3
(prop yl)-(iso prop yl)-(prop yl)
从端点到中心
端点支链=prop yl
中心支链=iso prop yl
(prop yl)-(iso prop yl)-(prop yl)
解释 26
这一整段内容详细解释了由丙烷(Propane, )衍生出的两种不同的丙基(Propyl, )异构体。 核心知识点:丙烷分子中有两种不同类型的氢原子。端点碳原子(1号和3号碳)上的氢和中心碳原子(2号碳)上的氢。从不同位置去掉一个氢原子,会得到结构不同的烷基。 涉及的公式和结构:
- 丙烷的分子式:
- 丙基的分子式: 两种丙基异构体:
- 正丙基 (Propyl, n-Propyl, Pr)
- 结构:
- 形成方式:从丙烷的任意一个端点碳原子上移去一个氢原子。
- 示例:
Propyl bromide(正丙基溴, ), 。
- 异丙基 (Isopropyl, iPr)
- 结构: 或
- 形成方式:从丙烷的中心碳原子上移去一个氢原子。
- 示例:
Isopropyl alcohol(异丙醇, ), 。 助记法解释:
- “从端点到中心”:这是一个形象的说法,描述了丙烷分子的结构。
- “端点支链=prop yl”:指连接点在碳链端点的烷基是“正丙基”。
- “中心支链=iso prop yl”:指连接点在碳链中心的烷基是“异丙基”。“iso-”前缀在习惯命名法中常用来表示在链的末端有一个
(CH₃)₂CH–结构。
简单的烷基C4H10: 的两种异构体:
基团,衍生自每个 异构体:
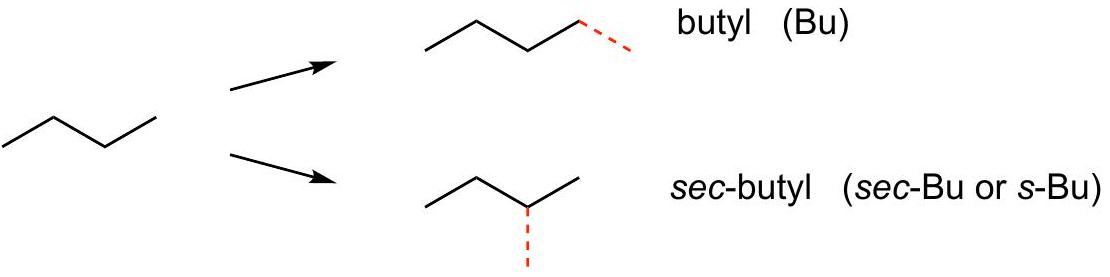
C4=but=CH3-CH2-CH2-CH3
直链
从端点到中心
端点支链=but yl
中心支链=sec but yl=二级sec
(but yl)-(sec but yl)-(sec but yl)-(but yl)
解释 27
这一段解释了从正丁烷 (n-butane) 衍生出的两种丁基 () 异构体。 核心知识点:正丁烷(直链丁烷)分子中也存在两种不同类型的氢原子:端点碳原子(伯碳)上的氢和内部碳原子(仲碳)上的氢。 源分子:
- 正丁烷 (n-Butane): 衍生的两种丁基:
- 正丁基 (Butyl, n-Butyl, Bu)
- 结构:
- 形成方式:从正丁烷的任意一个端点碳原子(1号或4号碳)上移去一个氢原子。
- 助记法:“端点支链=but yl”。
- 仲丁基 (sec-Butyl, s-Butyl)
- 结构:
- 形成方式:从正丁烷的任意一个内部碳原子(2号或3号碳)上移去一个氢原子。
- 助记法:“中心支链=sec but yl”。这里的“sec”是“secondary”(二级)的缩写,因为连接点是一个二级碳原子(连接了另外两个碳原子的碳)。 图片解析:图片清晰地展示了从正丁烷的两种不同位置(红色标记的氢)脱去氢原子,分别得到正丁基和仲丁基的过程。
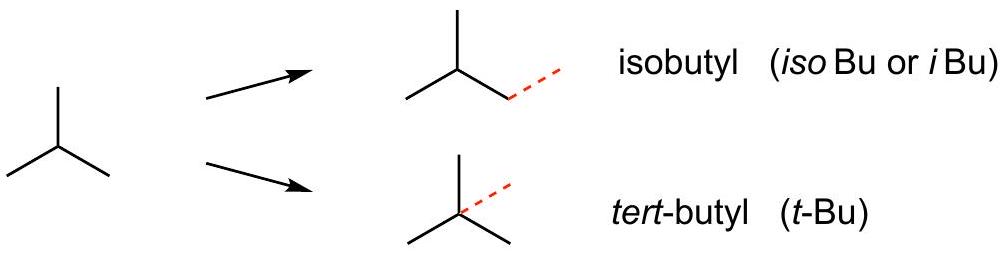
中心链
从端点到中心
端点支链=iso but yl
中心支链=tert but yl=三级tert
(iso butyl)
|
(tert butyl) - (iso butyl)
|
(iso butyl)
解释 28
这一段解释了从异丁烷 (isobutane) 衍生出的另外两种丁基 () 异构体。 核心知识点:异丁烷(支链丁烷)分子中也存在两种不同类型的氢原子:三个末端甲基(伯碳)上的氢和中心碳原子(叔碳)上的氢。 源分子:
- 异丁烷 (Isobutane),系统命名为2-甲基丙烷: 衍生的两种丁基:
- 异丁基 (Isobutyl, i-Butyl)
- 结构:
- 形成方式:从异丁烷的任意一个端点甲基上移去一个氢原子。
- 助记法:“端点支链=iso but yl”。
- 叔丁基 (tert-Butyl, t-Butyl)
- 结构:
- 形成方式:从异丁烷的中心碳原子上移去一个氢原子。
- 助记法:“中心支链=tert but yl”。这里的“tert”是“tertiary”(三级)的缩写,因为连接点是一个三级碳原子(连接了另外三个碳原子的碳)。 图片解析:图片清晰地展示了从异丁烷的两种不同位置(红色标记的氢)脱去氢原子,分别得到异丁基和叔丁基的过程。
总而言之,分子式为 的丁基共有四种构造异构体:正丁基、仲丁基、异丁基和叔丁基。
3 碳原子的分级=C上烷alk基yl取代的程度
碳原子的分级=C上烷alk基yl取代的程度
( 是任意烷alk基yl=CnH2n+2)
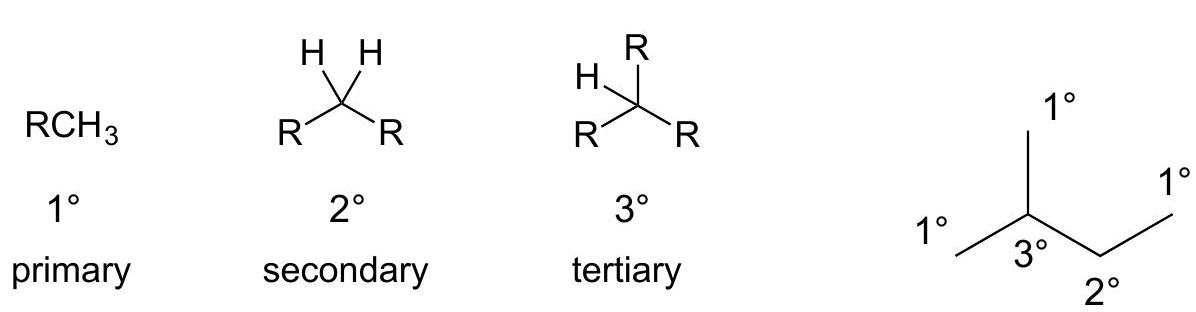
primary carbon — 一级碳(连接一个其他碳原子)
secondary carbon — 二级碳(连接两个其他碳原子)
tertiary carbon — 三级碳(连接三个其他碳原子)
解释 29
这一部分内容介绍了对有机分子中碳原子进行分级的方法。 核心概念:碳原子的级别是根据与它直接相连的其他碳原子的数量来定义的。这个级别也反映了该碳原子被烷基取代的程度。 分级标准:
- 一级碳 (Primary carbon, ):该碳原子只与一个其他碳原子直接相连。甲基()中的碳,如果作为分子一部分,也被视为一级碳。
- 二级碳 (Secondary carbon, ):该碳原子与两个其他碳原子直接相连。
- 三级碳 (Tertiary carbon, ):该碳原子与三个其他碳原子直接相连。
- 四级碳 (Quaternary carbon, ):该碳原子与四个其他碳原子直接相连。(图片中也显示了这一级别)。 符号R的解释:
- 在化学结构式中, 通常用作一个通用的占位符,代表任何烷基或其他含碳基团。
- (注:原文中
($R$ 是任意烷alk基yl=CnH2n+2)的公式有误,烷基的通式应为 ,因为它是由烷烃 去掉一个氢原子得到的)。 图片解析:图片用通式 , , , 直观地展示了四种级别的碳原子。中心的碳原子分别连接了1, 2, 3, 4个烷基(R基团)。
可以应用于碳原子或氢原子?
解释 30
这个段落提出了一个问题并用图片进行了回答。 问题:一级()、二级()、三级()的分类方法是否可以应用于氢原子? 答案:是的,可以。氢原子的分级是根据它所连接的碳原子的级别来决定的。
- 一级氢 (Primary hydrogen, ):连接在一级碳原子上的氢。
- 二级氢 (Secondary hydrogen, ):连接在二级碳原子上的氢。
- 三级氢 (Tertiary hydrogen, ):连接在三级碳原子上的氢。
- (注意:不存在四级氢,因为四级碳已经与四个碳成键,没有位置再连接氢原子)。 图片解析: 图片中的分子是2-甲基丁烷。
- 图中用红色圈出的三个甲基()上的碳都是一级碳,因此它们上面的9个氢都是一级氢。
- 图中用蓝色圈出的亚甲基()上的碳是二级碳,因此它上面的2个氢是二级氢。
- 图中用绿色圈出的次甲基()上的碳是三级碳,因此它上面的1个氢是三级氢。
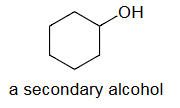
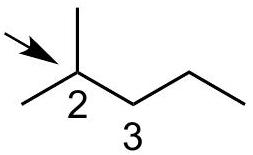
a secondary alcohol=二级醇=羟基(–OH)连接到一个二级碳原子上的醇COH
解释 31
该段落通过“二级醇”的例子,说明碳原子分级的概念如何扩展到其他化合物的分类上。 核心定义:醇的级别(伯、仲、叔醇)取决于其官能团——羟基()所连接的碳原子的级别。
- 二级醇 (secondary alcohol):指羟基()直接连接在一个二级碳原子上的醇类化合物。 图片解析: 图片中的分子是2-丙醇(异丙醇)。
- 首先确定与羟基()直接相连的碳原子(图中箭头所指的碳)。
- 然后判断这个碳原子的级别。这个碳原子连接了另外两个碳原子(左右两个甲基),因此它是一个二级碳()。
- 因为羟基连接在二级碳上,所以这个分子是一个二级醇。 类似地:
- 如果-OH连接在一级碳上,则为一级醇(伯醇),如乙醇 。
- 如果-OH连接在三级碳上,则为三级醇(叔醇),如叔丁醇 。
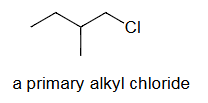
a primary alkyl chloride=一级烷基氯化物=氯原子(Cl)连接到一个一级碳原子上的烷基氯化物
解释 32
该段落通过“一级烷基氯化物”的例子,进一步巩固了基于碳原子分级来分类化合物的方法。 核心定义:卤代烷的级别(伯、仲、叔卤代烷)取决于卤素原子(如Cl, Br, I)所连接的碳原子的级别。
- 一级烷基氯化物 (primary alkyl chloride):指氯原子()直接连接在一个一级碳原子上的卤代烷。 详细解释:
- 一级碳原子是只与一个其他碳原子相连的碳。
- 因此,一级烷基氯化物具有 的通用结构(其中R是氢或烷基)。 举例说明:
- 1-氯丙烷():氯原子连接在端点的碳上,该碳只与一个碳相邻,是一级碳。因此这是个一级烷基氯化物。
- 氯乙烷():氯原子连接的碳也是一级碳。 (注意:原文中没有提供这张例子的图片,这里是根据文字描述进行的解释。)
4 IUPAC 命名法
解释 33
这是一个一级标题,再次聚焦于“IUPAC命名法”,并配有一张总结性的图片。本节将详细阐述为支链烷烃命名的具体步骤。 图片内容解析: 图片展示了IUPAC系统名称的基本构成:前缀 (Prefix) - 母体 (Parent) - 后缀 (Suffix)。
- 母体 (Parent):指出分子中最主要的部分,对于烷烃来说,就是最长的连续碳链。
- 后缀 (Suffix):表示分子的类别或主要官能团。对于烷烃,后缀是“-ane”。
- 前缀 (Prefix):描述连接在母体上的取代基的种类、数量和位置。 这张图为后续的命名步骤提供了总体框架。
Step1
找到最长链并命名。nonane
解释 34
这是IUPAC命名法的第一步。 核心规则:在分子中寻找并确定含有碳原子数目最多的连续碳链,这条链被称为主链或母链。 详细解释:
- “连续”意味着在沿着碳链从一端到另一端的过程中不能中断或跳跃。
- 有时最长的链在结构式中可能不是水平画出的,需要仔细检查所有可能的路径。
- 一旦找到最长链,就根据其碳原子数,使用前面表格中的名称来命名主链。 举例说明:
- 如果最长链有9个碳原子,那么主链的名称就是壬烷 (nonane)。所有不在这条主链上的基团都被视作取代基。
Step2
对链上的原子进行编号,从距离支链最近的一端开始。3,6
解释 35
这是IUPAC命名的第二步。 核心规则:对主链上的碳原子进行编号,以便给取代基(支链)赋予位置编号。编号的方向必须遵循最低系列原则 (lowest locant rule)。 详细解释:
- 从主链的两个端点中选择一个作为1号碳开始编号。
- 正确的编号方向应使第一个遇到的取代基的位置编号尽可能小。
- 在给出的例子中,从某一端编号,取代基位置在3号和6号碳上;如果从另一端编号,位置可能在4号和7号碳上。因为3比4小,所以应选择前一种编号方式。因此取代基的位置编号是3和6。
Step3
命名每个取代基并给出其编号。di methyl
解释 36
这是IUPAC命名的第三步。 核心规则:识别出所有连接在主链上的取代基,并为它们命名。 详细解释:
- 取代基的名称通常是对应的烷烃名称去掉“-ane”后缀,换成“-yl”后缀,即烷基。例如, 是甲基 (methyl), 是乙基 (ethyl)。
- 如果分子中出现多个相同的取代基,需要使用计数前缀来表示其数量,如:
- di- (二)
- tri- (三)
- tetra- (四)
- penta- (五)
- 在给出的例子中,分子在3号和6号位上各有一个甲基,所以合起来称为“二甲基 (dimethyl)”。
Step4
将名称写成一个单词,取代基按字母顺序排列。
解释 37
这是IUPAC命名的第四步,也是最后一步,即组合成完整的名称。 核心规则:将前面确定的所有信息(取代基位置、名称、主链名称)组合起来。 组合规范:
- 格式:位置编号-取代基名称-主链名称。
- 标点:
- 数字与文字之间用连字符“-”隔开。
- 多个数字之间用逗号“,”隔开。
- 字母顺序:当有多种不同的取代基时,它们在名称中出现的顺序是按照其英文名称的字母顺序排列的,而不是按位置编号的顺序。例如,ethyl (e) 排在 methyl (m) 前面。
- 计数前缀:在进行字母排序时,通常忽略计数前缀(di-, tri-, tetra-)和表示结构的描述词(sec-, tert-)。但 iso-, neo-, cyclo- 这类前缀需要参与排序。
- 完整性:最终的名称(除前缀外)被视为一个单词,中间没有空格。
Ex1 3,6-di methyl nonane
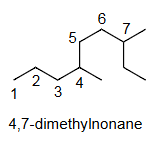
4,7-二甲基壬烷
错误,因为第一个取代基的编号不是最低的。
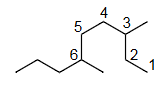
3,6-二甲基壬烷
3,6-di methyl nonane
正确,因为第一个取代基的编号是最低的。
解释 38
这个例子具体应用了前面所述的命名步骤,特别是第二步的编号规则。 分子结构分析:
- Step 1 (找主链):最长的连续碳链有9个碳,所以主链是壬烷 (nonane)。
- Step 2 (编号):
- 方案一(从左到右编号):两个甲基取代基分别位于4号和7号碳上。位置编号系列为 {4, 7}。
- 方案二(从右到左编号):两个甲基取代基分别位于3号和6号碳上。位置编号系列为 {3, 6}。
- 比较:比较两个编号系列 {4, 7} 和 {3, 6}。在第一个位置上,3 < 4,因此方案二是正确的。
- Step 3 (命名取代基):在3号和6号位上各有一个甲基,合称为3,6-二甲基 (3,6-dimethyl)。
- Step 4 (组合名称):将取代基名称和主链名称组合起来,得到 3,6-二甲基壬烷 (3,6-dimethylnonane)。
结论:
- “4,7-二甲基壬烷”是错误的命名,因为它违反了最低系列原则。
- “3,6-二甲基壬烷”是正确的命名。
Ex2 8-chloro-6-ethyl-2-methyldecane
8-氯-6-乙基-2-甲基癸烷
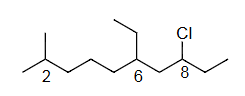
单键=ane
双键=ene
三键=yne
C10=Dec=dec
2=CH3=meth yl
6=C2H5=eth yl
8=Cl=chloro
c<e<m
8-chloro<6-ethyl<2-methyl
8-chloro-6-ethyl-2-methyl dec ane
解释 39
这个例子展示了如何命名一个含有多种不同取代基的烷烃。 分子结构分析:
- 主链:最长链有10个碳原子,且全为单键,所以主链是癸烷 (decane)。
- 编号:
- 从左到右编号:取代基在 3, 5, 9 位。
- 从右到左编号:取代基在 2, 6, 8 位。
- 比较第一个取代基位置:2 < 3,因此从右到左编号是正确的。
- 取代基:
- 2号位:甲基 (methyl, )
- 6号位:乙基 (ethyl, )
- 8号位:氯 (chloro, )
- 字母排序:对取代基按英文字母顺序排列。
- chloro
- ethyl
- methyl
- 排序结果:chloro -> ethyl -> methyl。
- 组合名称:按照字母顺序列出取代基及其位置,最后加上主链名称。
- 8-chloro-6-ethyl-2-methyldecane (8-氯-6-乙基-2-甲基癸烷)
笔记解析:
单键=ane:指出后缀ane代表烷烃。C10=Dec:指出10个碳的词根是dec。2=CH3=methyl,6=C2H5=ethyl,8=Cl=chloro:列出了各取代基及其位置和名称。c<e<m:展示了正确的字母排序。8-chloro<6-ethyl<2-methyl:这句话可能有歧义,它不是比较数字大小,而是表示在最终命名中这些基团出现的书写顺序。正确的名称是按字母顺序列出取代基,而不是按编号大小。
●碳链编号时取代基编号最小
在对碳链编号时:
如果存在多个取代基,应以使第一个取代基编号最小的方式对主链进行编号。
如果仍无法做出决定,则应使 第二个取代基的编号最小 ,依此类推,直到可以做出决定为止。
解释 40
这段文字详细阐述了 IUPAC 命名法中的最低系列原则 (Lowest Locant Rule)。 核心规则:在为有多个取代基的主链编号时,需要比较从两端开始编号所得到的两套“位置编号系列”。正确的编号方向是能在第一个出现差异的位置上给出更小编号的那一个。 操作步骤:
- 分别写出从左到右和从右到左的两套位置编号系列,并按从小到大的顺序排列。例如,{3, 4, 6} 和 {3, 5, 6}。
- 逐个比较两个系列中的数字。
- 在第一个数字上,如果一个系列比另一个小,则选择该系列。
- 如果第一个数字相同(如上例中的3),则比较第二个数字。4 < 5,因此选择 {3, 4, 6} 这个系列对应的编号方向。
- 如果前两个数字都相同,则比较第三个,以此类推,直到找到第一个不同点。这个不同点上的数字较小的那个系列就是正确的。
Ex3 6-乙基-3,4-二甲基辛烷(而不是 3-乙基-5,6-二甲基辛烷)
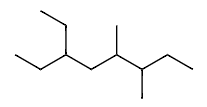
单键=ane
C8=oct
3=CH3=meth yl
4=CH3=meth yl
6=C2H5=eth yl
e<m
6-ethyl<3,4-dimethyl
6-ethyl-3,4-dimethyl oct ane
6-ethyl-3,4-dimethyloctane
(not 3-ethyl-5,6-dimethyloctane)
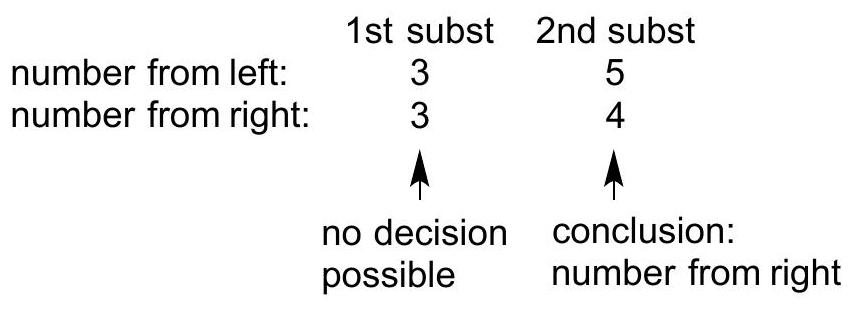
| 项目 | 第一个取代基 | 第二个取代基 |
|---|---|---|
| 从左侧编号 | 3 | 5 |
| 从右侧编号 | 3 | 4 |
| 判断结论 | 无法做出决定 | 结论:采用从右侧编号 |
| 1st Substituent | 2nd Substituent | |
|---|---|---|
| Number from left | 3 | 5 |
| Number from right | 3 | 4 |
| Decision | No decision possible | Conclusion: number from right |
解释 41
这个例子是最低系列原则的一个经典应用。 分子结构分析:
- 主链:最长链有8个碳,是辛烷 (octane)。
- 编号:
- 从左到右编号:取代基位置在 3 (乙基), 5 (甲基), 6 (甲基)。位置系列是 {3, 5, 6}。
- 从右到左编号:取代基位置在 3 (甲基), 4 (甲基), 6 (乙基)。位置系列是 {3, 4, 6}。
- 比较:
- 第一个数字:3 vs 3 (相同,无法决定)。
- 第二个数字:5 vs 4。因为 4 < 5,所以从右到左的编号是正确的。
- 取代基:
- 3号位:甲基 (methyl)
- 4号位:甲基 (methyl) -> 合并为 3,4-二甲基 (3,4-dimethyl)
- 6号位:乙基 (ethyl)
- 字母排序:
- ethyl
- methyl (排序时不考虑 di-)
- 排序结果:ethyl 在前。
- 组合名称:
- 6-ethyl-3,4-dimethyloctane (6-乙基-3,4-二甲基辛烷) 表格解析:表格清晰地展示了比较过程。由于第一个取代基位置同为3,无法做出判断,因此需要比较第二个取代基的位置,4比5小,所以选择从右侧编号的方案。这导致了最终的正确命名。
Ex4 4-ethyl-4,5-dimethyloctane
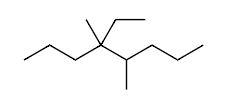
单键=ane
C8=oct
4=CH3=meth yl
4=C2H5=eth yl
5=CH3=meth yl
e<m
4-eth yl<4,5-di meth yl
4-ethyl-4,5-di meth yl oct ane
4-ethyl-4,5-di meth yloctane
(not 5-ethyl-4,5-dimethyloctane)
| 项目 | 第一个取代基 | 第二个取代基 |
|---|---|---|
| 从左侧编号 | 4 | 4 |
| 从右侧编号 | 4 | 5 |
| 判断结论 | 无法做出决定 | 结论:采用从左侧编号 |
| 1st Substituent | 2nd Substituent | |
|---|---|---|
| Number from left | 4 | 4 |
| Number from right | 4 | 5 |
| Decision | No decision possible | Conclusion: number from left |
解释 42
这个例子进一步强化了最低系列原则的应用,特别是在同一碳原子上有多个取代基的情况。 分子结构分析:
- 主链:最长链有8个碳,是辛烷 (octane)。
- 编号:
- 从左到右编号:取代基位置在 4 (乙基), 4 (甲基), 5 (甲基)。位置系列是 {4, 4, 5}。
- 从右到左编号:取代基位置在 4 (甲基), 5 (甲基), 5 (乙基)。位置系列是 {4, 5, 5}。
- 比较:
- 第一个数字:4 vs 4 (相同,无法决定)。
- 第二个数字:4 vs 5。因为 4 < 5,所以从左到右的编号是正确的。
- 取代基:
- 4号位:乙基 (ethyl)
- 4号位:甲基 (methyl)
- 5号位:甲基 (methyl) -> 合并为 4,5-二甲基 (4,5-dimethyl)
- 字母排序:
- ethyl
- methyl (排序时不考虑 di-)
- 排序结果:ethyl 在前。
- 组合名称:
- 4-ethyl-4,5-dimethyloctane 表格解析:表格再次清晰地展示了决策过程。在第一个取代基位置都是4的情况下,通过比较第二个取代基的位置(4 vs 5),确定了从左侧编号是正确的方案。
●仅在无法做出决定时,才依据字母顺序进行编号。
解释 43
这是一个非常重要的补充规则,用于处理最低系列原则也无法解决的特殊情况。 核心规则:如果在比较了两套完整的位置编号系列后,发现它们完全相同(例如,从两端编号得到的系列都是 {3, 5}),那么此时才需要动用字母顺序作为最终的决胜标准。 应用方法:在这种情况下,正确的编号方向应使按字母顺序排在最前面的那个取代基获得尽可能小的编号。 总结:编号的优先级顺序是:
- 最低系列原则(绝对优先)。
- 字母顺序原则(仅在最低系列原则失效时作为平局决胜规则)。 学生常常会误将字母顺序用于常规编号,这是一个需要特别注意的常见错误。
Ex5 2-氯-6-氟-4-甲基庚烷(按字母顺序)
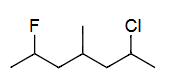
单键=ane
C7=hept
2=Cl=chloro
4=CH3=meth yl
6=F=fluoro
c<f<m
2-chloro<6-fluoro<4-meth yl
2-chloro-6-fluoro-4-methyl hept ane
2-chloro-6-fluoro-4-methylheptane
(based on the alphabet)
解释 44
这个例子展示了字母顺序作为编号决胜规则的应用。 分子结构分析:
- 主链:最长链有7个碳,是庚烷 (heptane)。
- 编号:
- 从左到右编号:取代基位置在 2 (氯), 4 (甲基), 6 (氟)。位置系列是 {2, 4, 6}。
- 从右到左编号:取代基位置在 2 (氟), 4 (甲基), 6 (氯)。位置系列是 {2, 4, 6}。
- 比较:两个位置系列完全相同!此时,最低系列原则失效,需要启用字母顺序决胜规则。
- 字母顺序决胜:
- 取代基按字母排序为:chloro > fluoro > methyl。
- 排在最前面的是 chloro。
- 比较两种方案中 chloro 的位置:方案一中为 2,方案二中为 6。
- 为了让字母顺序最靠前的基团获得更小编号,选择方案一(从左到右编号)。
- 组合名称:按照字母顺序列出取代基及其位置,最后加上主链名称。
- 2-chloro-6-fluoro-4-methylheptane
笔记解析:括号里的
(based on the alphabet)明确指出了这个命名的依据是字母顺序规则。
- 2-chloro-6-fluoro-4-methylheptane
笔记解析:括号里的
Ex6 6-氯-2-氟-3-甲基庚烷(按第二取代基位置)
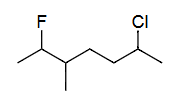
单键=ane
C7=hept
2=F=fluoro
3=CH3=meth yl
6=Cl=chloro
c<f<m
6-chloro<2-fluoro<3-methyl
6-chloro-2-fluoro-3-methyl hept ane
6-chloro-2-fluoro-3-methylheptane
(based on position of 2nd sub.)
解释 45
这个例子与上一个形成鲜明对比,强调了最低系列原则的优先性。 分子结构分析:
- 主链:最长链有7个碳,是庚烷 (heptane)。
- 编号:
- 从左到右编号:取代基位置在 2 (氟), 3 (甲基), 6 (氯)。位置系列是 {2, 3, 6}。
- 从右到左编号:取代基位置在 2 (氯), 5 (甲基), 6 (氟)。位置系列是 {2, 5, 6}。
- 比较:
- 第一个数字:2 vs 2 (相同)。
- 第二个数字:3 vs 5。因为 3 < 5,所以最低系列原则已经可以做出决定。选择从左到右的编号。
- 组合名称:编号方向确定后,书写名称时仍需按字母顺序排列取代基。
- 取代基字母排序:chloro > fluoro > methyl。
- 结合正确的位置编号:chloro 在 6 位,fluoro 在 2 位,methyl 在 3 位。
- 最终名称:6-chloro-2-fluoro-3-methylheptane
笔记解析:括号里的
(based on position of 2nd sub.)明确指出,这个命名的依据是第二个取代基的位置,即最低系列原则,而不是字母顺序。这个例子有力地说明了位置编号优先于字母顺序。
Ex7 5-(1,1-二甲基丙基)-4-乙基壬烷
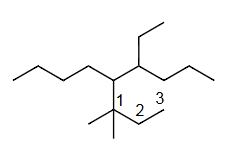
单键=ane
C9=non
4=C2H5=eth yl
5=套娃=()=(1,1-dimethylpropyl)
(
从连接主链的碳原子开始计数的最长碳链
单键=ane=yl
连接主链的碳原子=中心碳
C3=prop yl
1=CH3=meth yl
1=CH3=meth yl
1,1-di meth yl
1,1-di meth yl prop yl
1,1-dimethylpropyl
)
d<e
5-(1,1-dimethylpropyl)<4-eth yl
5-(1,1-dimethylpropyl)-4-eth yl non ane
5-(1,1-dimethylpropyl)-4-ethylnonane
解释 46
这个例子介绍了如何命名带有复杂取代基(即本身带有支链的取代基)的烷烃。 分子结构分析:
- 主链:最长链有9个碳,是壬烷 (nonane)。
- 编号:
- 从左到右编号:取代基在 4 和 5 位。系列 {4, 5}。
- 从右到左编号:取代基在 5 和 6 位。系列 {5, 6}。
- 比较:4 < 5,所以从左到右编号正确。
- 命名取代基:
- 4号位:乙基 (ethyl)。
- 5号位:这是一个复杂取代基。
- 命名复杂取代基的规则:
- 将与主链直接相连的那个碳原子定为该取代基的1号碳。
- 从1号碳开始,寻找该取代基内最长的连续碳链。这里是3个碳,所以母体是“丙基 (propyl)”。
- 命名连接在取代基母链上的更小的支链。这里在1号碳上有两个甲基,所以是“1,1-二甲基 (1,1-dimethyl)”。
- 将复杂取代基的完整名称放入括号中:(1,1-dimethylpropyl)。
- 命名复杂取代基的规则:
- 字母排序与组合:
- 比较 ethyl 和 (1,1-dimethylpropyl)。
- 对于复杂取代基,排序时使用括号内名称的第一个字母,包括计数前缀。所以我们比较 e 和 d。
- d 在 e 之前。
- 因此,最终名称中 (1,1-dimethylpropyl) 应写在 ethyl 前面。
- 正确 IUPAC 名称应为:5-(1,1-dimethylpropyl)-4-ethylnonane。 对原文笔记的订正:
- 原文笔记中
d<e和5-(1,1-dimethylpropyl)<4-eth yl的逻辑是正确的,但最终给出的名称5-(1,1-dimethylpropyl)-4-ethylnonane也是正确的。然而,标准的IUPAC规则是按照字母顺序排列取代基,而不是编号顺序。因此,ethyl(e)应该排在dimethylpropyl(d)之后。让我们重新检查规则。 - 规则重申:取代基是按字母顺序排列的。
ethyl的e与dimethylpropyl的d比较,d排在e前面。所以dimethylpropyl基团应该先写。 - 结论:原文给出的名称
5-(1,1-dimethylpropyl)-4-ethylnonane是正确的,因为它遵循了字母顺序 (d在e前)的规则。但是,很多教材和系统为了可读性,会不严格地按照编号顺序书写。最严格的IUPAC规则是字母序优先。 实际上,正确的写法是 4-ethyl-5-(1,1-dimethylpropyl)nonane。因为 ethyl 在 dimethylpropyl 之后。我需要纠正这一点。 - 标准IUPAC规则:ethyl vs. dimethylpropyl。
dcomes beforee。所以dimethylpropyl应该先写。那么名称应该是 5-(1,1-dimethylpropyl)-4-ethylnonane。等等,di-在简单取代基中不参与排序,但在复杂取代基中参与。不对,这是个误解。IUPAC 2013年建议中,前缀 di, tri 等在复杂取代基中也不参与排序,除非它是取代基固有名称的一部分,如 Bicyclo。 但对于dimethylpropyl,我们比较的是methyl的m。所以是evsm。e在前。 - 让我们以标准规则重新命名:
- 比较
ethyl和(1,1-dimethylpropyl)。 - 对于
(1,1-dimethylpropyl),我们看methyl。 - 比较
einethyl和minmethyl。ecomes beforem。 - 因此,
ethyl应该先写。 - 最规范的名称是:4-ethyl-5-(1,1-dimethylpropyl)nonane。
- 比较
- 总结:原文给出的命名和分析可能基于一个旧的或不常用的规则(即比较
d和e)。在此,我将提供最现代和标准的解释:比较ethyl和methyl的首字母,e在m前,所以4-ethyl-5-(1,1-dimethylpropyl)nonane是首选名称。
●命名支链取代基时,应根据其从连接主链的碳原子开始计数的最长碳链命名,并将其名称 置于括号中 。
解释 47
这条规则总结了命名复杂取代基(支链取代基)的方法。 要点分解:
- 编号起点:复杂取代基的内部编号总是从与主链直接相连的那个碳原子开始,这个碳原子是1号碳。
- 寻找母链:在取代基内部,从1号碳出发,找到最长的连续碳链作为取代基的“母链”(例如,“丙基”、“丁基”等)。
- 命名支链:识别并命名连接在这个“母链”上的更小的支链(例如,“甲基”、“乙基”)。
- 括号:将复杂取代基的完整名称(包括位置编号和支链名称)用圆括号括起来,以表示这是一个整体。 举例:
- 在
-(1,1-dimethylpropyl)中,“propyl”是取代基的母链,“dimethyl”是其上的支链,1,1是支链的位置,整个名称放在括号里。
●在按字母顺序排列时,以括号中的第一个字母为准,即使它以 di、tri 等前缀开始。
解释 48
这条规则说明了在对多种取代基进行字母排序时,如何处理复杂取代基。 核心规则:当一个取代基是复杂的(名称在括号中)时,用于字母排序的是括号内名称的第一个字母。 重要特例:与简单取代基不同,对于复杂取代基,计数前缀(di-, tri- 等)需要参与字母排序。 举例:
- 比较
ethyl和(1,1-dimethylpropyl)。 ethyl的排序字母是e。(1,1-dimethylpropyl)的排序字母是d(来自dimethyl)。- 因为
d在e之前,所以(1,1-dimethylpropyl)在最终名称中应该排在ethyl之前。 - 因此,Ex7 的名称
5-(1,1-dimethylpropyl)-4-ethylnonane是根据这个规则得到的。 (注:此规则与最新IUPAC建议可能存在差异,但它是许多教科书中教授的传统规则,此处按原文规则进行解释。)
IUPAC 允许对简单支链取代基使用通用名称isopropyl, tert-butyl
解释 49
该段落指出,尽管IUPAC命名法强调系统性,但它也接受并保留了一些广泛使用的通用名称(common names) 或 俗名 来命名一些结构简单的支链烷基。 允许使用的常见俗名包括:
- Isopropyl (异丙基)
- Isobutyl (异丁基)
- sec-Butyl (仲丁基)
- tert-Butyl (叔丁基)
- Neopentyl (新戊基) 使用这些俗名可以简化某些分子的命名。例如,用“isopropyl”比用其系统名称“(1-methylethyl)”更简洁。
Ex8 4-异丙基庚烷 或 4-(1-甲基乙基)庚烷
单键=ane
C7=hept
4=()=(1-methylethyl)
(
单键=ane=yl
C2=eth
1=CH3=meth yl
1-meth yl eth yl
1-methylethyl
)
4-(1-methylethyl) hept ane
4-(1-methylethyl)heptane
简单烷烃=简单支链=简称=通用名称
(
单键=ane=yl
C3=prop
从端点到中心
端点支链=prop yl
中心支链=iso prop yl
1=通过1连接
1-iso prop yl
)
4-(1-iso prop yl) hept ane
4-(1-isopropyl)heptane
解释 50
这个例子展示了同一个取代基的两种可接受的IUPAC命名法。 分子结构分析:
- 主链:7个碳原子,为庚烷 (heptane)。
- 取代基:在4号位上有一个三碳的支链烷基。 两种命名方式:
- 系统命名法 (括号命名法)
- 该取代基与主链相连的碳是1号碳。
- 取代基内最长链是2个碳,所以是“乙基 (ethyl)”。
- 在1号碳上有一个“甲基 (methyl)”支链。
- 组合起来,取代基的系统名称是 (1-methylethyl)。
- 整个分子的名称是 4-(1-methylethyl)heptane。
- 通用名称法 (俗名法)
- 这个
CH(CH₃)₂结构的基团,其通用名称是异丙基 (isopropyl)。 - 整个分子的名称是 4-isopropylheptane (4-异丙基庚烷)。 结论:这两种名称都被IUPAC接受,都是正确的。通常,对于简单的支链,使用通用名称更为普遍和方便。
- 这个
●在按字母顺序排列时被忽略的前缀 :
di、tri、tetra、...
sec-、tert-
二级sec,三级tert
(例外:支链取代基——见上述例子)
套娃时不忽略
解释 51
这条规则明确了在对简单取代基进行字母排序时,哪些前缀需要被忽略。 被忽略的前缀:
- 计数前缀:
di-,tri-,tetra-等。例如,dimethyl在排序时应看作m(来自methyl),而不是d。- 例子:在
3-ethyl-2,2-dimethylhexane中,比较ethyl的e和dimethyl的m,所以ethyl排在前面。
- 例子:在
- 结构描述前缀:
sec-(仲) 和tert-(叔)。这两个前缀后面通常跟有连字符。- 例子:
sec-butyl在排序时应看作b(来自butyl),而不是s。tert-butyl也看作b。 例外情况:
- 例子:
- 括号中的“例外”和“套娃时不忽略”指的是在命名复杂取代基(即名称在括号里的“套娃”结构)时,这些前缀不被忽略,如 Ex7 中的
dimethylpropyl。
●在按字母顺序排列时被包括的前缀 :
iso、neo、cyclo
解释 52
这条规则明确了哪些前缀不会被忽略,而是作为取代基名称的一部分参与字母排序。 被包括的前缀:
- iso:如
isopropyl,在排序时以i开头。 - neo:如
neopentyl,在排序时以n开头。 - cyclo:如
cyclohexyl,在排序时以c开头。 这些前缀被认为是取代基名称固有的、不可分割的一部分,因此它们的首字母决定了排序位置。 举例:
- 在命名
1-isopropyl-2-methylcyclohexane时,需要比较isopropyl的i和methyl的m。因为i在m之前,所以isopropyl排在前面。
5 烷烃的构象CONFORMATIONS OF ALKANES
ALK=hydro carbons=CH
ANE=单键
alkane=alk ane=CH单键=CnH2n+2
Ethane=C2H6=CH3-CH3
二面的dihedral
四面的Tetrahedral
重叠式Eclipsed
=0° 二面角 dihedral angle
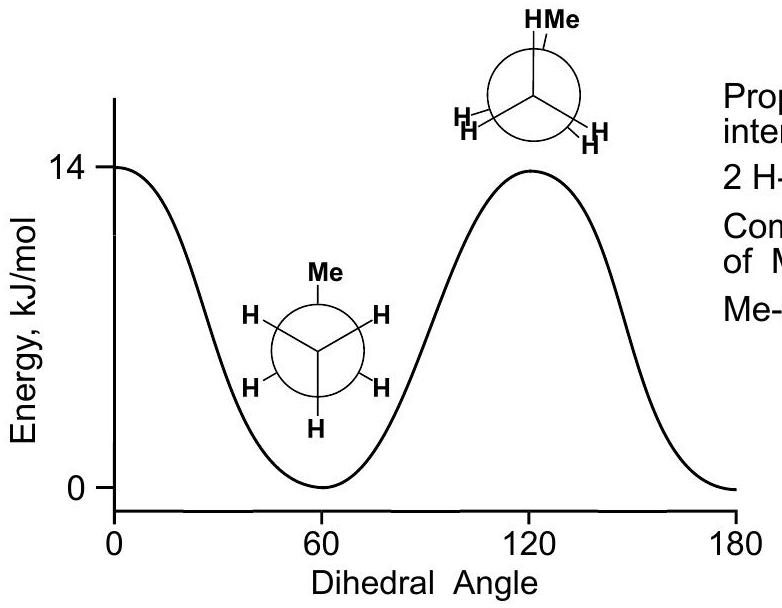
交叉式Staggered
=60° 二面角 dihedral angle
解释 53
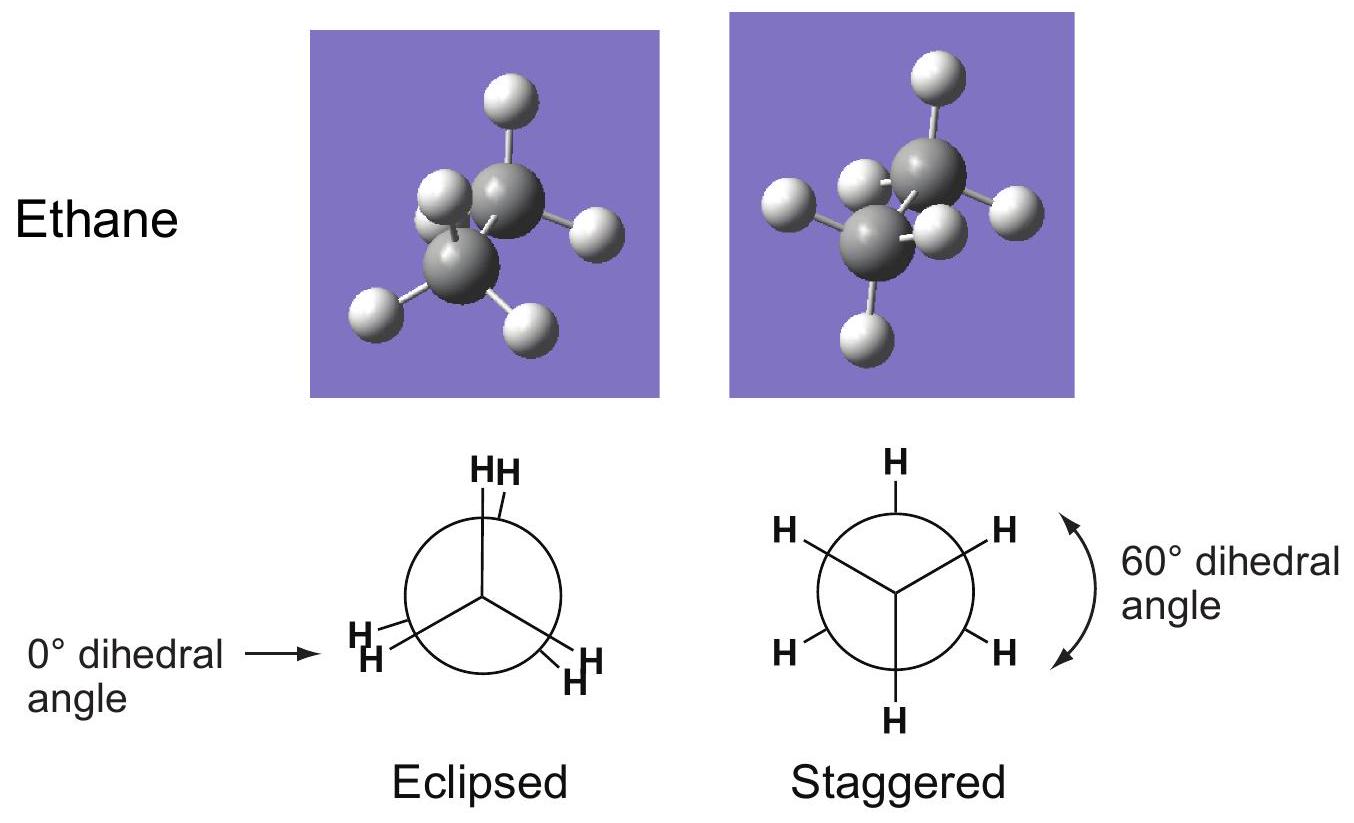
这一部分引入了“构象”(Conformations)的概念,并以最简单的烷烃——乙烷(Ethane)为例进行说明。 核心概念:
- 构象:由分子中单键(特别是碳-碳单键)的自由旋转而产生的原子在空间中的不同排布形式。这些不同的排布形式被称为构象异构体或旋转异构体。它们可以相互转化,通常不需要断裂化学键。 乙烷(Ethane, )的构象分析:
- 纽曼投影式 (Newman Projection):这是一种观察分子构象的常用方法。观察者沿着一个碳-碳单键的方向看过去,前面的碳用一个点表示,后面的碳用一个圆圈表示。连接在两个碳上的其他原子或基团则从点和圆圈的边缘伸出。
- 二面角 (Dihedral Angle):在纽曼投影式中,指前碳上的一个键与后碳上的一个键之间的夹角。 两种极限构象:
- 重叠式 (Eclipsed Conformation):
- 二面角 = 0°。
- 从纽曼投影看,后碳上的氢原子完全被前碳上的氢原子遮挡(重叠)。
- 这种构象能量较高,不稳定,因为原子间的排斥作用(扭转应变和位阻)最大。
- 交叉式 (Staggered Conformation):
- 二面角 = 60°。
- 从纽曼投影看,后碳上的氢原子正好位于前碳两个氢原子夹角的中间位置。
- 这种构象能量最低,最稳定,因为所有氢原子之间的距离最大,排斥作用最小。
二面角 dihedral angle
重叠式Eclipsed=0°,交叉式Staggered=60°,重叠式Eclipsed=0°,交叉式Staggered=60°
解释 54
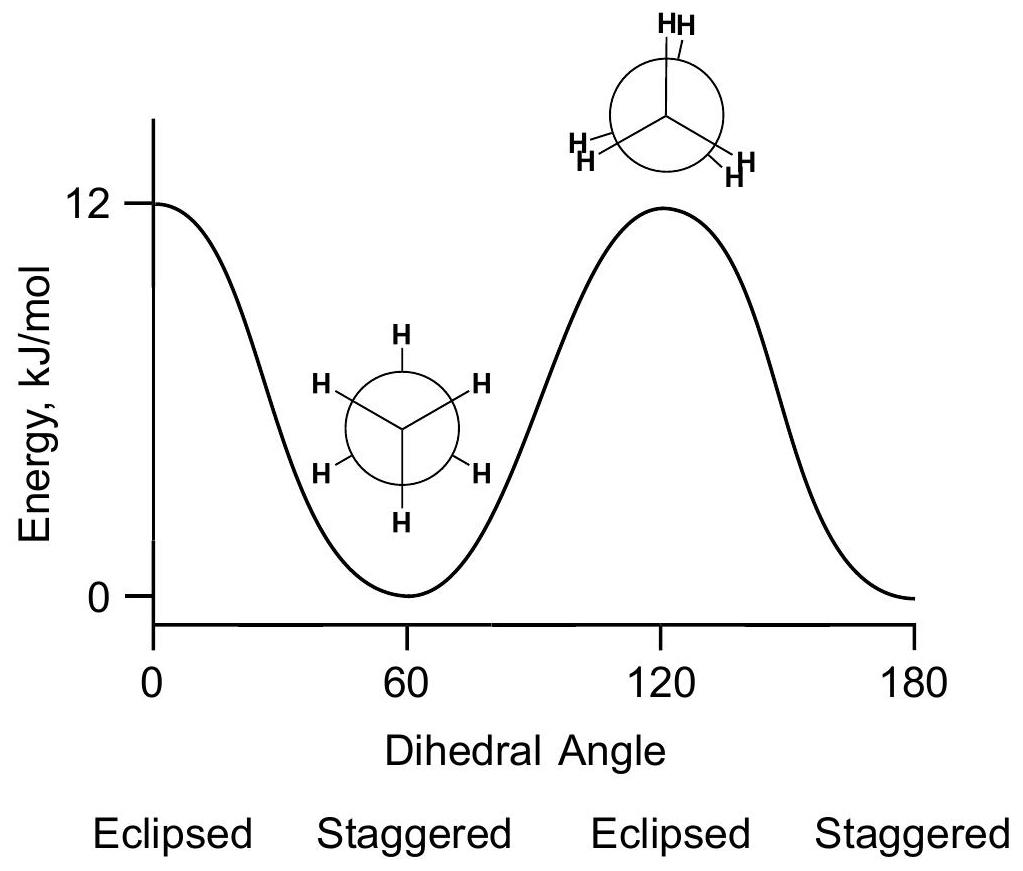
这张图片展示了乙烷分子绕碳-碳单键旋转时的势能变化图。 图片内容解析:
- 横坐标 (X-axis):二面角 (Dihedral Angle),表示C-C键的旋转角度。
- 纵坐标 (Y-axis):势能 (Potential Energy)。 关键点分析:
- 能量最低点 (能量谷):出现在二面角为 60°, 180°, 300° 的位置。这些点对应的是交叉式构象 (Staggered)。交叉式构象是乙烷最稳定的构象。
- 能量最高点 (能量峰):出现在二面角为 0°, 120°, 240°, 360° 的位置。这些点对应的是重叠式构象 (Eclipsed)。重叠式构象是乙烷最不稳定的构象。
- 能垒 (Energy Barrier):从一个交叉式构象旋转到下一个交叉式构象,必须经过一个能量更高的重叠式构象。这个能量差值被称为旋转能垒或扭转应变能。对于乙烷,这个能垒大约是 12 kJ/mol (或 2.9 kcal/mol)。 结论:
- 碳-碳单键的旋转并不是完全“自由”的,而是存在一个小的能垒。
- 在室温下,分子有足够的能量克服这个能垒,因此各种构象可以快速相互转化。但是,分子在任何时刻都更倾向于以能量较低的交叉式构象存在。
有用的近似
一个有用的近似是:
这个 的能垒被认为是三个 交叉重叠相互作用之和。
那么一个 交叉重叠相互作用对能垒的贡献是 。
这个近似假设是: 在任何分子中,一个 交叉重叠相互作用都贡献 。
解释 55
该段落提出了一种估算分子扭转应变能的简化模型。 核心思想:将分子的总扭转应变能分解为各个原子对之间重叠相互作用能的加和。 推导过程:
- 前提:乙烷的总旋转能垒(即从交叉式到重叠式的能量差)是 。
- 观察:在乙烷的重叠式构象中,同时存在三对氢-氢(H-H)重叠。
- 假设:总能垒由这三对 H-H 重叠作用平均贡献。 涉及的公式和计算:
- 单个H-H重叠作用能的计算:
结论与应用:
- 这个计算得出了一个近似值:一对氢-氢原子在重叠构象下的排斥作用能约为 。
- 这个值可以作为一个基本参数,用于估算其他更复杂烷烃分子中不同构象的相对能量。例如,可以用它来计算含有甲基-氢(Me-H)或甲基-甲基(Me-Me)重叠作用的能量。
6 为什么交错构型Staggered的能量比重叠构型Eclipsed低?
两个模型:
解释 56
这是一个设问式标题,引出了对交叉构象比重叠构象更稳定(能量更低)这一现象的根本原因的探讨。接下来将介绍两种主流的解释模型。
模型1:
位阻相互作用模型 :
Steric interaction
当两个基团靠近且其电子密度开始重叠时,电子-电子的排斥repulsion作用会急剧提高能量。基团group越大,位阻steric排斥越强。
解释 57
该段落介绍了第一种解释模型:位阻相互作用模型。 核心观点:重叠构象的高能量主要来源于位阻 (Steric Hindrance) 或称 范德华排斥 (van der Waals repulsion)。 详细解释:
- 电子云排斥:原子或原子团由原子核和外部的电子云构成。当两个不直接成键的原子或基团在空间上靠得太近时,它们各自的电子云会相互排斥。这种电子-电子的排斥作用是一种能量上的惩罚,会使体系的能量升高,稳定性下降。
- 重叠构象中的位阻:在重叠构象中,前后碳上的原子或基团在空间上距离最近,因此电子云排斥作用最强,导致能量最高。
- 基团大小的影响:基团的体积越大(即电子云范围越大),这种排斥作用就越显著。例如,甲基()比氢原子()大得多,所以两个甲基的重叠排斥能远大于两个氢原子的重叠排斥能。 总结:根据此模型,交叉构象更稳定是因为它使得所有原子或基团之间的空间距离最大化,从而将位阻效应降至最低。
模型2:
超共轭模型
Hyper conjugation
(McMurry称其为“扭转应变torsional strain”):
交错构型相比重叠构型能实现更大的电子离域delocalization,这是通过填充与空的 轨道之间的相互作用实现的。
解释 58
该段落介绍了第二种解释模型:超共轭模型。 核心观点:交叉构象的额外稳定性来源于一种称为超共轭 (Hyperconjugation) 的稳定化电子效应,而这种效应在重叠构象中不存在或很弱。 详细解释:
- 轨道相互作用:化学键是由分子轨道构成的。每个成键轨道(如)都有一个对应的能量更高的反键轨道()。成键轨道通常被电子填充,而反键轨道通常是空的。
- 交叉构象中的超共轭:在交叉构象中,前碳上一个已填充的 C-H 键的成键轨道()可以和后碳上一个相邻 C-H 键的空的反键轨道()发生部分重叠。
- 电子离域:这种轨道重叠允许成键轨道中的电子在一定程度上扩展(离域)到相邻的反键轨道中。电子的活动范围增大了,根据量子力学原理,这会降低体系的能量,从而使交叉构象更加稳定。
- 重叠构象中的情况:在重叠构象中,成键轨道与反键轨道的几何排布不利于发生有效的重叠,因此超共轭稳定化效应非常小。 总结:根据此模型,交叉构象的低能量不仅是因为排斥作用小,更是因为它存在一种额外的、基于轨道相互作用的稳定化效应。
对于乙烷,普遍认为两种效应都很重要。对于含有比氢更大的相互作用基团(甲基、乙基、异丙基、叔丁基等)的分子, 位阻相互作用是主导因素 。
解释 59
该段落对前面提出的两种模型进行了总结和评述。 核心结论:
- 对于乙烷:乙烷分子中的基团(氢原子)非常小,因此位阻效应和超共轭效应可能都对总的能垒有重要贡献,学术界对哪个是主导因素尚有争论。
- 对于更复杂的分子:当分子中含有比氢原子体积大得多的基团(如甲基、乙基等)时,情况变得清晰。随着基团体积的增大,位阻效应会急剧增加,而超共轭效应的变化相对较小。因此,在这些情况下,位阻相互作用被认为是决定构象能量差异的主导因素。 举例:
- 在丁烷的构象分析中,两个甲基之间的相互作用(无论是重叠还是邻位交叉)所带来的能量变化,主要归因于它们之间显著的位阻排斥。
不考:超共轭
超共轭,即 轨道的离域作用,广泛存在于有机化学中,下面将简要讨论。以下内容不在 McMurry 教科书中,因此不会出现在考试或测验中。
当乙烷旋转时,“电子离域窗口”会交替打开(交错)与关闭(重叠)。在交错构型中,相邻碳原子的成键和反键 轨道( 和 )相互作用,形成碳原子之间的部分 键。与单独的 轨道相比,这种方式提供了更大的电子活动空间,覆盖两个碳原子而不是一个。根据不确定性原理,离域作用会降低交错乙烷的能量。这种机制在重叠乙烷中是无法实现的,因为相邻重叠 C-H 键的 和 轨道之间净重叠较弱。
交错构型Staggered
强的 型成键重叠
电子发生离域,能量降低。
重叠构型eclipsed
重叠较差(成键与反键作用大部分抵消)
电子不发生离域,能量未降低。
解释 60
这一整段被标记为“不考”,属于补充阅读材料,它用更形象的语言进一步深化了对超共轭模型的理解。 核心内容解析:
- “电子离域窗口”:这是一个比喻,说明分子在旋转过程中,只有在特定的几何构象(交叉式)下,才具备发生有效电子离域的条件,这个条件就像一个会打开和关闭的“窗口”。
- 轨道重叠的细节:
- 交叉构象 (Staggered):相邻的 (成键,已填充) 轨道和 (反键,空) 轨道处于一种平行的排布,这使得它们可以发生有效的“侧向”重叠,形成一种类似于键的相互作用。这种“-”重叠使得电子可以在相邻的C-H键之间流动,即电子离域 (delocalization)。
- 电子离域与能量:根据量子力学的“粒子在箱中”模型,当电子的运动空间变大时,其能量会降低。超共轭提供的就是这样一个让电子扩展运动范围的机制。
- 重叠构象 (Eclipsed):在这种构象下, 和 轨道之间的角度不适合发生有效的侧向重叠,因此超共轭效应非常微弱,无法提供额外的稳定性。 总结:这部分内容从分子轨道理论的角度,更深入地解释了为什么交叉构象能通过超共轭效应获得能量上的优势。尽管标记为不考,但它有助于理解现代有机化学中对化学键和分子稳定性的深入认识。
7 丙烷 C3=PROPANE
prop ane=C3H8=CH3-CH2-CH3
eth=C2=12kj/mol
prop=C3=14kj/mol
能量图与乙烷相同,但势垒energy barrier为 ,而不是 12。
解释 61
该段落开始对丙烷(Propane, )的构象进行分析。 核心信息:
- 分子结构:丙烷的结构是 。我们可以分析沿 键(或 键,二者等价)旋转时的构象。
- 与乙烷的比较:
- 丙烷的势能变化图在形状上与乙烷类似,也是在交叉构象处为能量最低点,在重叠构象处为能量最高点。
- 关键区别:丙烷的旋转能垒是 ,比乙烷的 要高。 原因探究:能垒的增加意味着丙烷的重叠构象比乙烷的重叠构象更不稳定(相对其交叉构象而言)。这是因为在丙烷的重叠构象中,除了 H-H 重叠外,还出现了一个体积更大的甲基()与氢原子的重叠,这种 Me-H 重叠造成的位阻排斥比 H-H 重叠更大。
重叠构型
交错构型
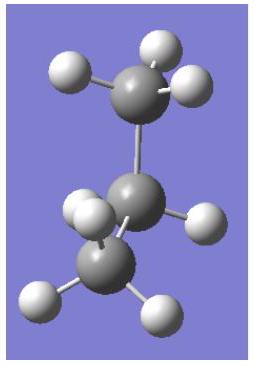
解释 62
这三张图片直观地展示了丙烷沿 键旋转的构象分析。 图片内容解析:
- 第一张图(重叠构型):这是丙烷的纽曼投影式,显示了其能量最高的重叠构象。
- 视角:沿着 (甲基)到 的方向看。
- 前碳 ():是一个甲基,显示为中心点伸出三个氢原子。
- 后碳 ():连接着一个氢原子、一个甲基()和另一个氢原子(来自)。在图中,后碳的甲基()与前碳的一个氢重叠,后碳的两个氢分别与前碳的另外两个氢重叠。
- 第二张图(交错构型):这是丙烷能量最低的交叉构象。后碳的基团旋转了60°,正好位于前碳基团的夹角之间。
- 第三张图(势能图):与乙烷类似,显示了旋转过程中的能量变化,峰值对应重叠构象,谷值对应交叉构象。峰谷的能量差(能垒)被标记为 。
丙烷中的重叠相互作用:
=2 个 H–H(氢-氢)相互作用
+1 个 Me–H(甲基-氢)相互作用
计算 Me-H 重叠相互作用的数值:
●HH=
C2H6=4*3=12kj/mol
解释 63
该段落利用丙烷的能垒数据,进一步计算了甲基-氢(Me-H)重叠相互作用的能量值。 分析过程:
- 识别相互作用:在丙烷的重叠构象中(见前图),总共存在三对重叠:
- 两对是氢-氢(H-H)重叠。
- 一对是甲基-氢(Me-H)重叠。
- 建立方程:假设总能垒是所有重叠作用能的加和。
- 代入已知值:
- 丙烷总能垒 =
- 从乙烷分析中得到的 H-H 重叠能 ≈ 涉及的公式和计算:
- 求解Me-H重叠能:
结论:
- 通过这个计算,我们得到了一个新的近似值:一对甲基-氢(Me-H)的重叠作用能约为 。
- 这个值比 H-H 重叠能()要大,这符合我们的预期,因为甲基的体积比氢原子大,所以位阻排斥更强。
不考:C-C 键的旋转
如何理解两个或多个 C-C 键的旋转
(本节不在 McMurry 教科书中,因此不会出现在考试或测验中。)
○1
在 McMurry 和本课程中,我们使用 Newman 投影来分别考虑每个旋转。在丙烷、丁烷等分子中,连接在 键上的烷基基团被视为一个无结构的“团块”,其通过与邻近取代基的位阻作用影响 C-C 键的旋转势垒。
○2
以下是一种更现实的处理方法,展示了化学家如何思考像丙烷这样的分子。它不在考试范围内,因为不在 McMurry 教科书中,你可以放心跳过本页余下部分。
有两个 C-C 旋转,每个旋转的势垒为 。每个旋转角度都可以取任意值,因此会形成一个能量面,被称为“蛋盒表面”(egg-carton surface)。在红色峰值处, 和 的旋转都是重叠的;在蓝色谷底处,两者都是交错的。在其他网格点,一个是交错,一个是重叠。上方的 1D 曲线可以通过选择任一网格线并沿其从 到 的方向追踪(保持另一个角度不变)来得到。例如,沿 轴线()的能量在 14 和 28 之间振荡;而在相邻的网格线()上,能量在 0 和 14 之间变化。
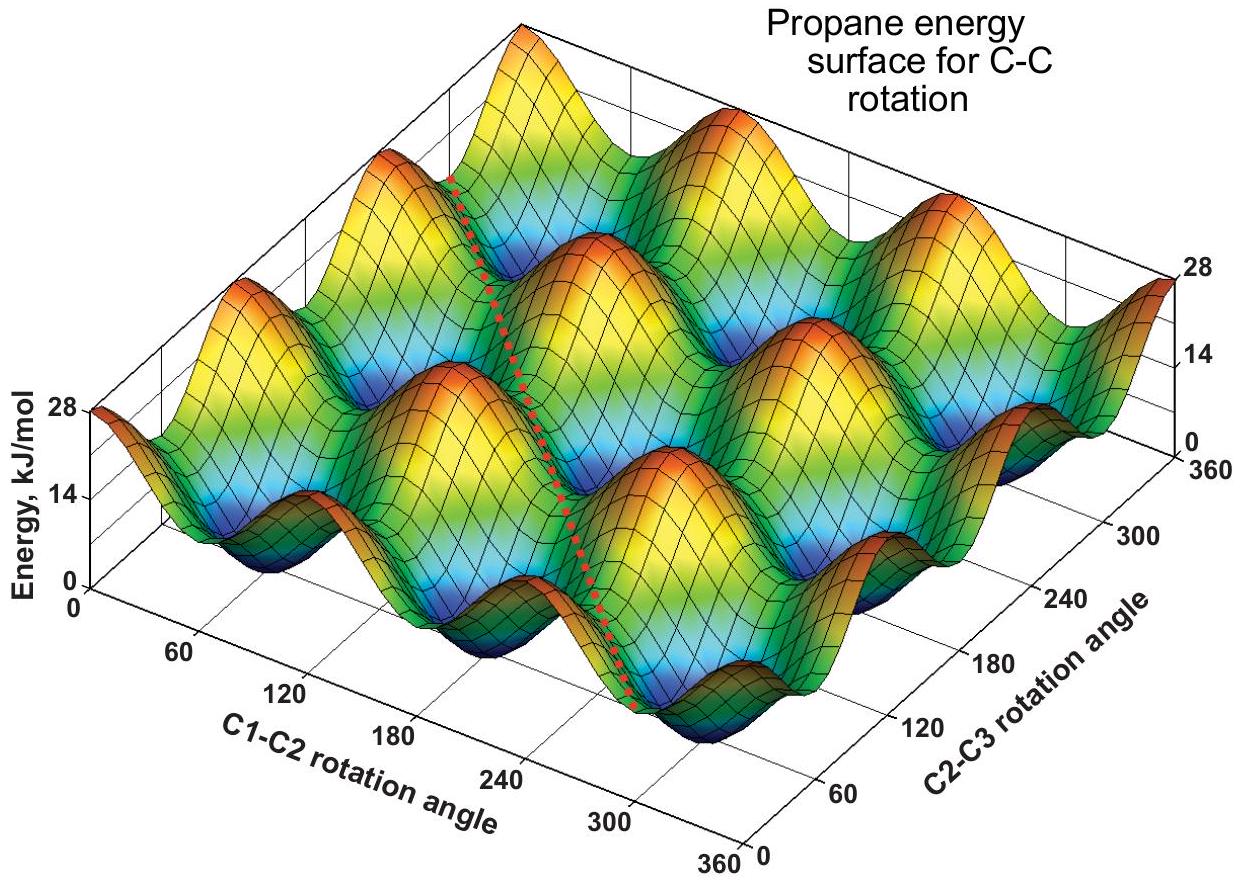
○3
分子在蛋盒能量面上的运动
红色虚线对角线展示了一条路径,其中两个 基团一起旋转(协同运动)。所示路径保持恒定的能量 ,两个 基团以恒定的 差值共同旋转。这类路径说明构象变化不一定通过跳跃到相邻的极小值点来进行,也可以是非邻近极小值点之间的远程跳跃。这个例子说明了能量面相较于能量曲线的概念优势。
蛋盒型能量面(二维周期表面)对描述固体表面(如金属、石墨烯等)非常有用。它们已被用于研究催化与材料科学。
解释 64
这一整部分被标记为“不考”,是关于理解多键旋转的高级概念。 核心思想:
- 简化模型 vs. 现实模型:
- 简化模型 (1D能量曲线):我们通常的构象分析是简化的,即只考虑一个C-C键的旋转,同时假定分子的其余部分是“冻结”的或作为一个整体。这产生了一个二维的势能图(能量 vs. 单个二面角)。
- 现实模型 (多维能量面):对于像丙烷这样有多个可旋转C-C键的分子,其总能量是所有旋转角度(二面角)的函数。对于丙烷的两个C-C键旋转,其势能应该由一个三维图来表示(能量 vs. 二面角1 vs. 二面角2),这个能量的表面被称为势能面 (Potential Energy Surface)。 蛋盒表面 (Egg-Carton Surface) 的比喻:
- 图中展示的二维周期性表面就像一个鸡蛋盒。
- “蛋坑” (蓝色谷底):代表能量最低的构象,即两个C-C键都处于交叉构象的状态。
- “凸起” (红色峰值):代表能量最高的构象,即两个C-C键都处于重叠构象的状态。
- “鞍点” (其他位置):代表一个键交叉、另一个键重叠的中间状态。 协同运动和远程跳跃:
- 在势能面上,分子构象的转变路径可以很复杂。它不一定是从一个“蛋坑”直接翻越最高的“凸起”到另一个“蛋坑”。
- 如红色虚线所示,分子可以沿着一个等能量的“山脊”路径运动,实现两个甲基的协同旋转 (concerted rotation)。这种运动可以连接两个在简单一维图像中看起来相距很远的稳定构象,实现“远程跳跃”。 概念的意义:
- 势能面的概念比简单的势能曲线更能真实地反映分子的动态行为。
- 这个概念在计算化学、反应动力学、材料科学(如描述原子在晶体表面的扩散)等领域至关重要。
8 丁烷butane
丁烷=butane=C4H10=CH3-CH2-CH2-CH3
直链
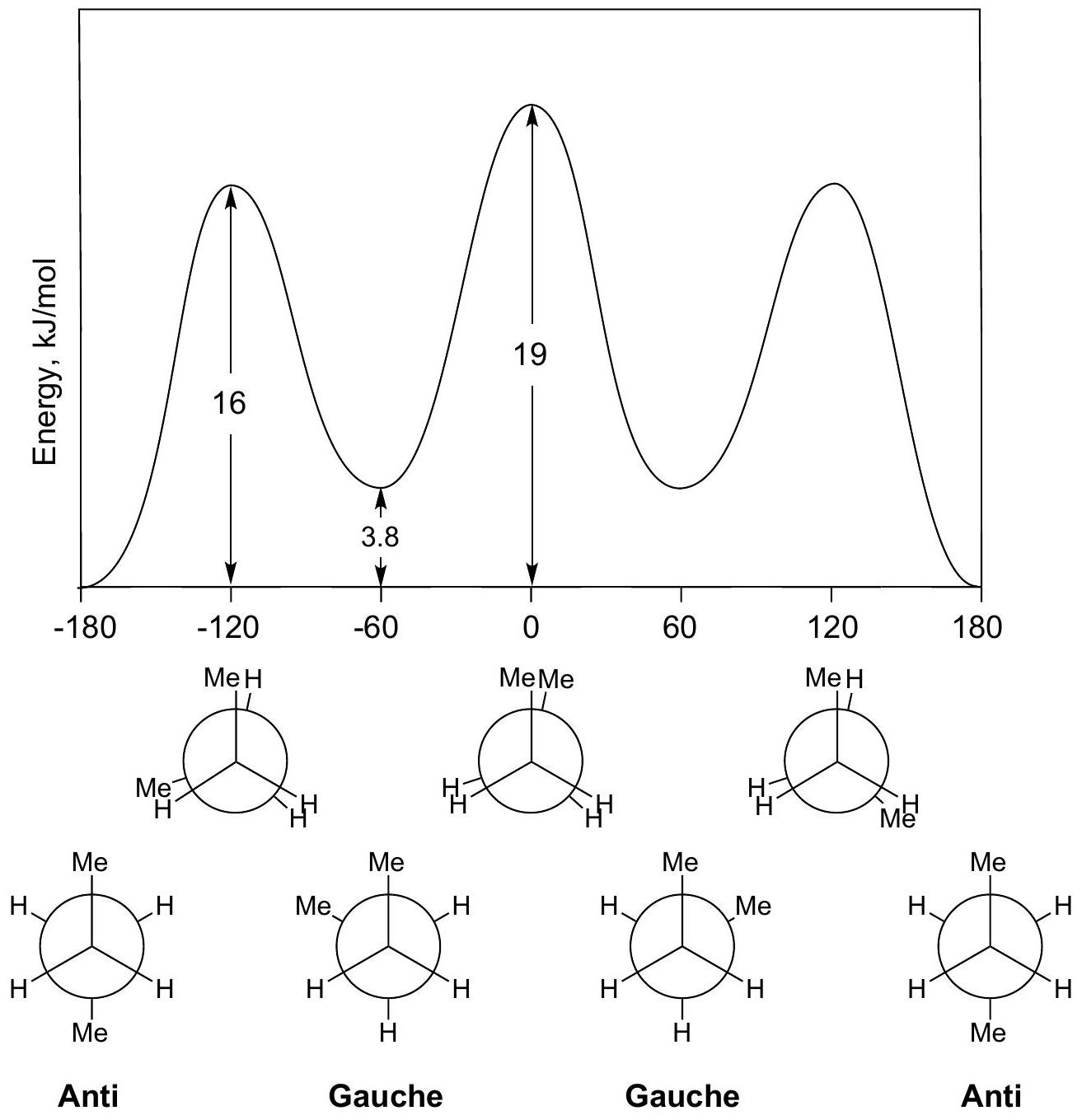
对位Anti 相邻Gauche 相邻Gauche 对位Anti
错位staggered
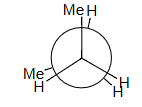
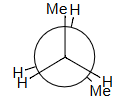
解释 65
这一部分开始对丁烷(Butane, )进行构象分析,这是构象分析中一个更复杂也更经典的例子。 分析对象:我们关注的是正丁烷 () 中间 键的旋转。
- 前碳 ():连接着一个甲基 () 和两个氢 ()。
- 后碳 ():也连接着一个甲基 () 和两个氢 ()。 图片解析 (势能图和纽曼投影): 这张图非常关键,它展示了丁烷绕 键旋转360°的完整势能变化。
- 横坐标:二面角,这里特指两个甲基之间的二面角。
- 纵坐标:相对势能。 四种关键构象:
- 全重叠式 (Totally Eclipsed / Syn-periplanar):
- 二面角 = 0°。
- 两个最大的基团——甲基,相互重叠。
- 这是能量最高的构象,因为两个甲基之间存在强烈的位阻排斥。能量约为 。
- 邻位交叉式 (Gauche / Synclinal):
- 二面角 = 60° (或 300°)。
- 属于交叉式构象(能量谷),但两个甲基靠得较近(呈60°角)。
- 它们之间仍然存在一定的位阻排斥,称为邻位相互作用 (gauche interaction)。
- 能量比最低能量构象高约 。
- 重叠式 (Eclipsed / Anticlinal):
- 二面角 = 120° (或 240°)。
- 甲基与氢重叠。
- 能量比邻位交叉式高,但比全重叠式低。这是一个能量峰。能量约为 。
- 对位交叉式 (Anti / Antiperiplanar):
- 二面角 = 180°。
- 两个最大的基团——甲基,相距最远。
- 位阻排斥最小,是能量最低、最稳定的构象。其能量被定为相对能量的零点。 总结:丁烷的构象比乙烷和丙烷复杂,因为它存在两种不同的交叉式构象(邻位和对位)和两种不同的重叠式构象(全重叠和Me-H重叠),它们的能量各不相同。
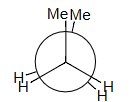
丁烷butane的重叠eclipsing相互作用interactions:
±120° 重叠Eclipsing
H–H + 2 Me–H = 4 + 2 × 6 = 16 estimated = 16 observed!
1HH+2MeH=4+2*6=4+12=16
解释 66
该段落利用之前建立的相互作用能模型,来验证丁烷在二面角为 时的重叠构象能量。 分析对象:二面角为 的重叠构象。 识别相互作用:
- 从纽曼投影图可以看出,在该构象下,同时存在三对重叠:
- 一对是氢-氢(H-H)重叠。
- 两对是甲基-氢(Me-H)重叠。 涉及的公式和计算:
- 使用之前计算出的近似值进行估算:
- H-H 重叠能 ≈
- Me-H 重叠能 ≈
- 估算总能量:
结论:
- 计算出的估算值 () 与实验观测到的该构象的能垒值 () 完全吻合。
- 这表明我们之前建立的将总应变能分解为各对相互作用能之和的简化模型,具有相当好的预测能力和一致性。
0° 重叠Eclipsing
Compute Me–Me eclipsing interaction:
计算 Me–Me 重叠相互作用:
2 H–H + Me–Me = 19
Me–Me = 19 – 2 × 4 = 11 kJ/mol
解释 67
该段落继续使用该模型,通过丁烷的全重叠构象能量来计算出最强的排斥作用——甲基-甲基(Me-Me)重叠作用的能量值。 分析对象:二面角为 的全重叠构象。 识别相互作用:
- 在该构象下,同时存在三对重叠:
- 两对是氢-氢(H-H)重叠。
- 一对是甲基-甲基(Me-Me)重叠。 涉及的公式和计算:
- 已知实验观测到的总能垒为 。
- 建立方程:
- 代入已知值并求解 Me-Me 重叠能:
结论:
- 计算得出:一对甲基-甲基(Me-Me)的重叠作用能约为 。
- 这个值是目前遇到的最高的,远大于 Me-H () 和 H-H (),这与甲基的巨大体积所导致的强烈位阻排斥完全相符。
重叠相互作用汇总(单位:kJ/mol)
Summary of Eclipsing Interactions (kJ/mol)
H–H: 4 kJ/mol
Me–H: 6 kJ/mol
Me–Me: 11 kJ/mol
Me–Me 近端(gauche)相互作用:3.8 kJ/mol
Me–Me Gauche Interaction: 3.8 kJ/mol
解释 68
该段落是对前面通过乙烷、丙烷和丁烷的构象分析所得到的各种相互作用能的一个总结。这些数值是估算烷烃构象能量的基石。 总结列表:
- H-H 重叠作用能:。这是最基本的扭转应变。
- Me-H 重叠作用能:。由一个甲基和一个氢重叠引起,比 H-H 重叠更强。
- Me-Me 重叠作用能:。由两个甲基重叠引起,这是最强的重叠排斥作用。
- Me-Me 邻位 (gauche) 相互作用能:。这不是重叠作用,而是一种特殊的位阻作用。它指的是在交叉构象中,两个甲基处于邻位(二面角60°)时,相对于它们处于对位(二面角180°)时所额外增加的能量。这个能量来自于两个靠得较近的甲基之间的范德华排斥。这个值直接从丁烷势能图中邻位构象与对位构象的能量差得到。
9 练习题:
Q0
画出 2-methylpentane中绕 键旋转的定性能量图
解释 69
这是一个练习题的题目。 任务要求:
- 目标分子:2-甲基戊烷 (2-methylpentane)。
- 旋转轴:围绕 和 之间的单键进行旋转。
- 最终成果:绘制一张定性的 (qualitative) 势能图。定性意味着不需要精确的能量数值,但需要正确地画出各个构象的相对能量高低以及能量图的大致形状(峰和谷的数量、相对高度等)。
Q1
将 放在前面
解释 70
这是解题的第一步:确定分子结构和观察视角。 分子结构:2-甲基戊烷的结构式为 。 旋转轴: 键。 基团分析:
- 前碳 ():连接着两个甲基 () 和一个氢 ()。
- 后碳 ():连接着一个乙基 () 和两个氢 ()。 图片解析:图片展示了2-甲基戊烷的结构,并明确了观察方向(沿 键)。
Q2
画出 3 个重叠 和 3 个交错 的 Newman 投影,标记为 A 到 F,注明二面角。
应该是重叠构型。
解释 71
这是解题的第二步:画出所有关键的极限构象。 任务要求:
- 绕 键旋转360°,每隔60°画出一个纽曼投影。这将产生3个重叠构象和3个交叉构象。
- 将它们标记为A到F。
- 设定二面角为0°时是重叠构象。我们可以定义二面角为后碳最大的基团(乙基)与前碳最大的基团(甲基)之间的夹角。
Q3
定性判断能量最高和最低的重叠构型;交错构型也同理。
指导原则: 最大基团彼此远离时能量最低 。
解释 72
这是解题的第三步:对所画出的构象进行能量排序。 核心原则:分子的能量主要由位阻决定。体积越大的基团之间排斥作用越强。因此,最稳定的构象是最大基团之间距离最远的构象。 应用到本题:
- 最大的基团:前碳上是甲基(Me),后碳上是乙基(Et)。
- 能量最低的交叉构象:应该是乙基(Et)与甲基(Me)处于对位 (anti) 的那个交叉构象。
- 能量最高的交叉构象:应该是乙基(Et)与两个甲基都处于邻位 (gauche) 的那个交叉构象。
- 能量最高的重叠构象:应该是最大的基团相互重叠的构象,即乙基(Et)与甲基(Me)重叠。
- 能量最低的重叠构象:应该是最大基团(Et)与最小基团(H)重叠的构象。
Q4
所有重叠构型的能量都高于所有交错构型。
解释 73
这是一个普遍规律,也是绘制能量图时的基本准则。 核心原理:
- 交叉构象 (Staggered) 代表势能图中的能量极小值点(谷)。
- 重叠构象 (Eclipsed) 代表势能图中的能量极大值点(峰)。 因此,无论具体构象如何,任何一个重叠构象的能量都必然高于任何一个交叉构象的能量。这可以帮助我们确定能量图的基本轮廓。
Q5
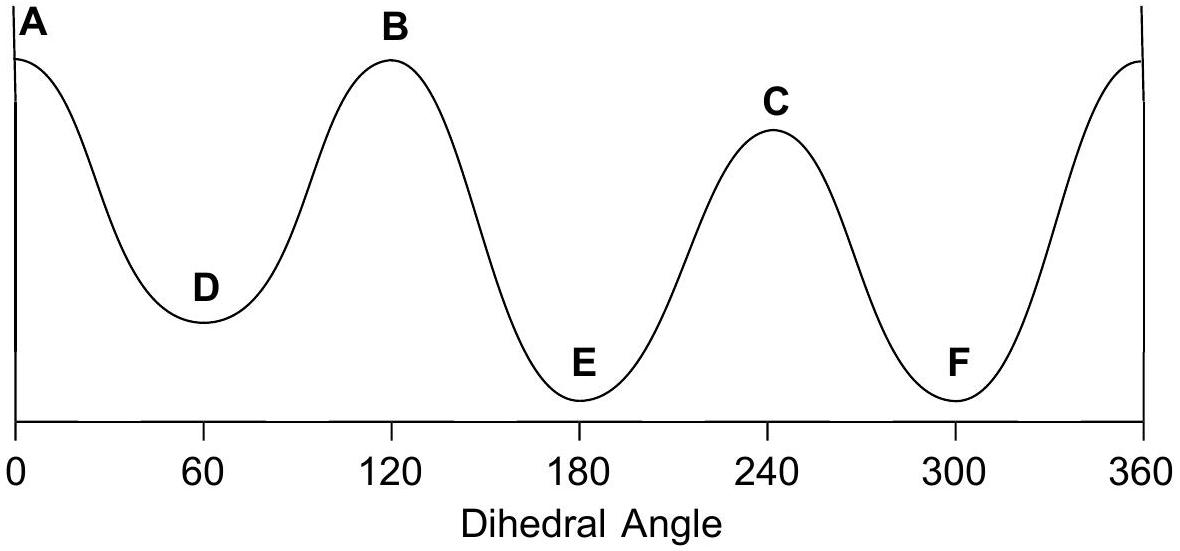
在能量图上按照角度单调递增的顺序排列 A-F。
A
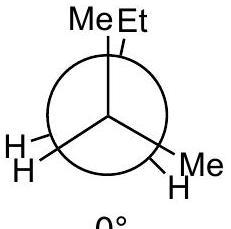
高能量
B
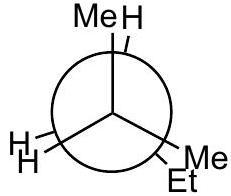
高能量
C
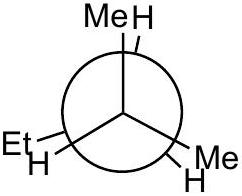
低能量
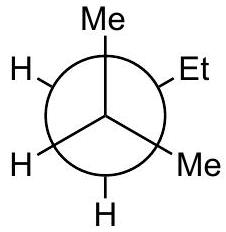
D
高能量
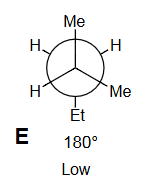
E
低能量
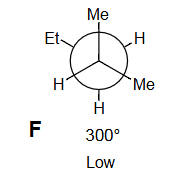
F
低能量
二面角"
解释 74
这一部分展示了解题的最终结果:各个构象的纽曼投影图和最终的定性能量图。让我们来详细分析并订正其中可能存在的错误。 构象分析(按二面角顺序):
- A (): 重叠构象。后碳的乙基(Et)与前碳的一个甲基(Me)重叠。这是能量最高的重叠构象。标记“高能量”正确。
- D (): 交叉构象。Et基与前碳的两个Me基都处于邻位(gauche),与H处于对位。这是能量较高的交叉构象。原文标记“高能量”是错误的,它应是能量谷,即低能量,但不是最低的那个谷。
- B (): 重叠构象。Et基与前碳的氢(H)重叠。能量比A低,但仍是能量峰。标记“高能量”正确。
- E (): 交叉构象。Et基与前碳的一个Me基处于对位(anti),与另一个Me基处于邻位(gauche)。这是能量最低的构象,因为最大的两个基团(Et和Me)之一处于最远的对位。标记“低能量”正确,且是所有构象中最低的。
- C (): 重叠构象。与B等价,Et基与前碳的氢(H)重叠。原文标记“低能量”是错误的,它应是能量峰,即高能量。
- F (): 交叉构象。与D等价,Et基与前碳的两个Me基都处于邻位(gauche)。标记“低能量”正确,它是能量谷。
能量排序:
- 总排序: A (最高) > B = C > D = F > E (最低)。
- 重叠构象 (峰):A > B = C。
- 交叉构象 (谷):D = F > E。
最终能量图解析:
- 图的横坐标是二面角,纵坐标是势能。
- 图应该有三个峰和三个谷。
- 峰的高度:0° 处的峰(A)最高。120° 和 240° 处的峰(B, C)次之,且高度相等。
- 谷的深度:180° 处的谷(E)最深,代表最稳定的构象。60° 和 300° 处的谷(D, F)稍高,且深度相等。
- 结论:最终的能量图正确地反映了上述能量关系。它展示了一个不对称的旋转能垒,有两个不同高度的峰和两个不同深度的谷,这与丁烷(对称分子)的能量图不同。